Coagulation
Coagulation, also known as clotting, is the process by which blood changes from a liquid to a gel, forming a blood clot. It potentially results in hemostasis, the cessation of blood loss from a damaged vessel, followed by repair. The mechanism of coagulation involves activation, adhesion and aggregation of platelets, as well as deposition and maturation of fibrin.
| Coagulation | |
|---|---|
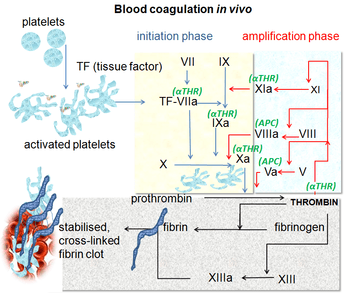 Blood coagulation pathways in vivo showing the central role played by thrombin | |
| Health | Beneficial |
Coagulation begins almost instantly after an injury to the blood vessel has damaged the endothelium lining the blood vessel. Exposure of blood to the subendothelial space initiates two processes: changes in platelets, and the exposure of subendothelial tissue factor to plasma factor VII, which ultimately leads to cross-linked fibrin formation. Platelets immediately form a plug at the site of injury; this is called primary hemostasis. Secondary . hemostasis occurs simultaneously: additional coagulation (clotting) factors beyond factor VII (listed below) respond in a cascade to form fibrin strands, which strengthen the platelet plug.[1]
Disorders of coagulation are disease states which can result in problems with hemorrhage, bruising, or thrombosis.[2]
Coagulation is highly conserved throughout biology. In all mammals, coagulation involves both a cellular (platelet) and a protein (coagulation factor) component.[3] The system in humans has been the most extensively researched and is the best understood.[4]
Physiology
Platelet activation
When the endothelium is damaged, the normally isolated, underlying collagen is exposed to circulating platelets, which bind directly to collagen with collagen-specific glycoprotein Ia/IIa surface receptors. This adhesion is strengthened further by von Willebrand factor (vWF), which is released from the endothelium and from platelets; vWF forms additional links between the platelets' glycoprotein Ib/IX/V and A1 domain. This localization of platelets to the extracellular matrix promotes collagen interaction with platelet glycoprotein VI. Binding of collagen to glycoprotein VI triggers a signaling cascade that results in activation of platelet integrins. Activated integrins mediate tight binding of platelets to the extracellular matrix. This process adheres platelets to the site of injury.[5]
Activated platelets release the contents of stored granules into the blood plasma. The granules include ADP, serotonin, platelet-activating factor (PAF), vWF, platelet factor 4, and thromboxane A2 (TXA2), which, in turn, activate additional platelets. The granules' contents activate a Gq-linked protein receptor cascade, resulting in increased calcium concentration in the platelets' cytosol. The calcium activates protein kinase C, which, in turn, activates phospholipase A2 (PLA2). PLA2 then modifies the integrin membrane glycoprotein IIb/IIIa, increasing its affinity to bind fibrinogen. The activated platelets change shape from spherical to stellate, and the fibrinogen cross-links with glycoprotein IIb/IIIa aid in aggregation of adjacent platelets (completing primary hemostasis).[6]
Coagulation cascade
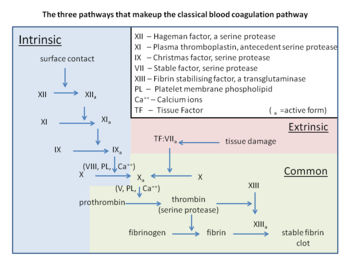
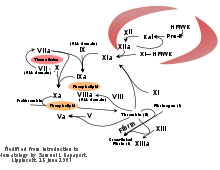
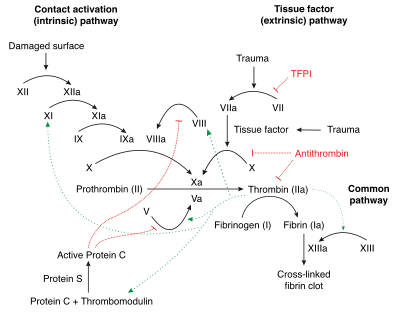
The coagulation cascade of secondary hemostasis has two initial pathways which lead to fibrin formation. These are the contact activation pathway (also known as the intrinsic pathway), and the tissue factor pathway (also known as the extrinsic pathway), which both lead to the same fundamental reactions that produce fibrin. It was previously thought that the two pathways of coagulation cascade were of equal importance, but it is now known that the primary pathway for the initiation of blood coagulation is the tissue factor (extrinsic) pathway. The pathways are a series of reactions, in which a zymogen (inactive enzyme precursor) of a serine protease and its glycoprotein co-factor are activated to become active components that then catalyze the next reaction in the cascade, ultimately resulting in cross-linked fibrin. Coagulation factors are generally indicated by Roman numerals, with a lowercase a appended to indicate an active form.[7]
The coagulation factors are generally serine proteases (enzymes), which act by cleaving downstream proteins. The exceptions are tissue factor, FV, FVIII, FXIII.[8] Tissue factor, FV and FVIII are glycoproteins, and Factor XIII is a transglutaminase.[7] The coagulation factors circulate as inactive zymogens. The coagulation cascade is therefore classically divided into three pathways. The tissue factor and contact activation pathways both activate the "final common pathway" of factor X, thrombin and fibrin.[9]
Tissue factor pathway (extrinsic)
The main role of the tissue factor pathway is to generate a "thrombin burst", a process by which thrombin, the most important constituent of the coagulation cascade in terms of its feedback activation roles, is released very rapidly. FVIIa circulates in a higher amount than any other activated coagulation factor. The process includes the following steps:[7]
- Following damage to the blood vessel, FVII leaves the circulation and comes into contact with tissue factor (TF) expressed on tissue-factor-bearing cells (stromal fibroblasts and leukocytes), forming an activated complex (TF-FVIIa).
- TF-FVIIa activates FIX and FX.
- FVII is itself activated by thrombin, FXIa, FXII and FXa.
- The activation of FX (to form FXa) by TF-FVIIa is almost immediately inhibited by tissue factor pathway inhibitor (TFPI).
- FXa and its co-factor FVa form the prothrombinase complex, which activates prothrombin to thrombin.
- Thrombin then activates other components of the coagulation cascade, including FV and FVIII (which forms a complex with FIX), and activates and releases FVIII from being bound to vWF.
- FVIIIa is the co-factor of FIXa, and together they form the "tenase" complex, which activates FX; and so the cycle continues. ("Tenase" is a contraction of "ten" and the suffix "-ase" used for enzymes.)
Contact activation pathway (intrinsic)
The contact activation pathway begins with formation of the primary complex on collagen by high-molecular-weight kininogen (HMWK), prekallikrein, and FXII (Hageman factor). Prekallikrein is converted to kallikrein and FXII becomes FXIIa. FXIIa converts FXI into FXIa. Factor XIa activates FIX, which with its co-factor FVIIIa form the tenase complex, which activates FX to FXa. The minor role that the contact activation pathway has in initiating clot formation can be illustrated by the fact that patients with severe deficiencies of FXII, HMWK, and prekallikrein do not have a bleeding disorder. Instead, contact activation system seems to be more involved in inflammation,[7] and innate immunity.[10] Despite this, interference with the pathway may confer protection against thrombosis without a significant bleeding risk.[10]
Final common pathway
The division of coagulation in two pathways is arbitrary, originating from laboratory tests in which clotting times were measured either after the clotting was initiated by glass, the intrinsic pathway; or clotting was initiated by thromboplastin (a mix of tissue factor and phospholipids), the extrinsic pathway.
Further, the final common pathway scheme implies that prothrombin is converted to thrombin only when acted upon by the intrinsic or extrinsic pathways, which is an oversimplification. In fact, thrombin is generated by activated platelets at the initiation of the platelet plug, which in turn promotes more platelet activation.
Thrombin functions not only to convert fibrinogen to fibrin, it also activates Factors VIII and V and their inhibitor protein C (in the presence of thrombomodulin); and it activates Factor XIII, which forms covalent bonds that crosslink the fibrin polymers that form from activated monomers.[7]
The coagulation cascade is maintained in a prothrombotic state by the continued activation of FVIII and FIX to form the tenase complex until it is down-regulated by the anticoagulant pathways.[7]
Cell-based scheme of coagulation
A newer model of coagulation mechanism explains the intricate combination of cellular and biochemical events that occur during the coagulation process in vivo. Along with the procoagulant and anticoagulant plasma proteins, normal physiologic coagulation requires the presence of two cell types for formation of coagulation complexes: cells that express tissue factor (usually extravascular) and platelets.
The coagulation process occurs in two phases. First is the initiation phase, which occurs in tissue-factor-expressing cells. This is followed by the propagation phase, which occurs on activated platelets. The initiation phase, mediated by the tissue factor exposure, proceeds via the classic extrinsic pathway and contributes to about 5% of thrombin production. The amplified production of thrombin occurs via the classic intrinsic pathway in the propagation phase; about 95% of thrombin generated will be during this second phase.[11]
Cofactors
Various substances are required for the proper functioning of the coagulation cascade:
Calcium and phospholipid
Calcium and phospholipid (a platelet membrane constituent) are required for the tenase and prothrombinase complexes to function. Calcium mediates the binding of the complexes via the terminal gamma-carboxy residues on FXa and FIXa to the phospholipid surfaces expressed by platelets, as well as procoagulant microparticles or microvesicles shed from them. Calcium is also required at other points in the coagulation cascade.
Vitamin K
Vitamin K is an essential factor to a hepatic gamma-glutamyl carboxylase that adds a carboxyl group to glutamic acid residues on factors II, VII, IX and X, as well as Protein S, Protein C and Protein Z. In adding the gamma-carboxyl group to glutamate residues on the immature clotting factors, Vitamin K is itself oxidized. Another enzyme, Vitamin K epoxide reductase (VKORC), reduces vitamin K back to its active form. Vitamin K epoxide reductase is pharmacologically important as a target of anticoagulant drugs warfarin and related coumarins such as acenocoumarol, phenprocoumon, and dicumarol. These drugs create a deficiency of reduced vitamin K by blocking VKORC, thereby inhibiting maturation of clotting factors. Vitamin K deficiency from other causes (e.g., in malabsorption) or impaired vitamin K metabolism in disease (e.g., in liver failure) lead to the formation of PIVKAs (proteins formed in vitamin K absence), which are partially or totally non-gamma carboxylated, affecting the coagulation factors' ability to bind to phospholipid.
Regulators
Five mechanisms keep platelet activation and the coagulation cascade in check. Abnormalities can lead to an increased tendency toward thrombosis:
Protein C
Protein C is a major physiological anticoagulant. It is a vitamin K-dependent serine protease enzyme that is activated by thrombin into activated protein C (APC). Protein C is activated in a sequence that starts with Protein C and thrombin binding to a cell surface protein thrombomodulin. Thrombomodulin binds these proteins in such a way that it activates Protein C. The activated form, along with protein S and a phospholipid as cofactors, degrades FVa and FVIIIa. Quantitative or qualitative deficiency of either (protein C or protein S) may lead to thrombophilia (a tendency to develop thrombosis). Impaired action of Protein C (activated Protein C resistance), for example by having the "Leiden" variant of Factor V or high levels of FVIII, also may lead to a thrombotic tendency.
Antithrombin
Antithrombin is a serine protease inhibitor (serpin) that degrades the serine proteases: thrombin, FIXa, FXa, FXIa, and FXIIa. It is constantly active, but its adhesion to these factors is increased by the presence of heparan sulfate (a glycosaminoglycan) or the administration of heparins (different heparinoids increase affinity to FXa, thrombin, or both). Quantitative or qualitative deficiency of antithrombin (inborn or acquired, e.g., in proteinuria) leads to thrombophilia.
Tissue factor pathway inhibitor (TFPI)
Tissue factor pathway inhibitor (TFPI) limits the action of tissue factor (TF). It also inhibits excessive TF-mediated activation of FVII and FX.
Plasmin
Plasmin is generated by proteolytic cleavage of plasminogen, a plasma protein synthesized in the liver. This cleavage is catalyzed by tissue plasminogen activator (t-PA), which is synthesized and secreted by endothelium. Plasmin proteolytically cleaves fibrin into fibrin degradation products that inhibit excessive fibrin formation.
Prostacyclin
Prostacyclin (PGI2) is released by endothelium and activates platelet Gs protein-linked receptors. This, in turn, activates adenylyl cyclase, which synthesizes cAMP. cAMP inhibits platelet activation by decreasing cytosolic levels of calcium and, by doing so, inhibits the release of granules that would lead to activation of additional platelets and the coagulation cascade.[12]
Fibrinolysis
Eventually, blood clots are reorganised and resorbed by a process termed fibrinolysis. The main enzyme responsible for this process (plasmin) is regulated by various activators and inhibitors.[12]
Role in immune system
The coagulation system overlaps with the immune system. Coagulation can physically trap invading microbes in blood clots. Also, some products of the coagulation system can contribute to the innate immune system by their ability to increase vascular permeability and act as chemotactic agents for phagocytic cells. In addition, some of the products of the coagulation system are directly antimicrobial. For example, beta-lysine, an amino acid produced by platelets during coagulation, can cause lysis of many Gram-positive bacteria by acting as a cationic detergent.[13] Many acute-phase proteins of inflammation are involved in the coagulation system. In addition, pathogenic bacteria may secrete agents that alter the coagulation system, e.g. coagulase and streptokinase.
Assessment
Numerous tests are used to assess the function of the coagulation system:[14]
- Common: aPTT, PT (also used to determine INR), fibrinogen testing (often by the Clauss method), platelet count, platelet function testing (often by PFA-100), thrombodynamics test.
- Other: TCT, bleeding time, mixing test (whether an abnormality corrects if the patient's plasma is mixed with normal plasma), coagulation factor assays, antiphospholipid antibodies, D-dimer, genetic tests (e.g. factor V Leiden, prothrombin mutation G20210A), dilute Russell's viper venom time (dRVVT), miscellaneous platelet function tests, thromboelastography (TEG or Sonoclot), euglobulin lysis time (ELT).
The contact activation (intrinsic) pathway is initiated by activation of the "contact factors" of plasma, and can be measured by the activated partial thromboplastin time (aPTT) test.
The tissue factor (extrinsic) pathway is initiated by release of tissue factor (a specific cellular lipoprotein), and can be measured by the prothrombin time (PT) test. PT results are often reported as ratio (INR value) to monitor dosing of oral anticoagulants such as warfarin.
The quantitative and qualitative screening of fibrinogen is measured by the thrombin clotting time (TCT). Measurement of the exact amount of fibrinogen present in the blood is generally done using the Clauss method for fibrinogen testing. Many analysers are capable of measuring a "derived fibrinogen" level from the graph of the Prothrombin time clot.
If a coagulation factor is part of the contact activation or tissue factor pathway, a deficiency of that factor will affect only one of the tests: Thus hemophilia A, a deficiency of factor VIII, which is part of the contact activation pathway, results in an abnormally prolonged aPTT test but a normal PT test. The exceptions are prothrombin, fibrinogen, and some variants of FX that can be detected only by either aPTT or PT. If an abnormal PT or aPTT is present, additional testing will occur to determine which (if any) factor is present as aberrant concentrations.
Deficiencies of fibrinogen (quantitative or qualitative) will affect all screening tests.
Role in disease
Coagulation defects may cause hemorrhage or thrombosis, and occasionally both, depending on the nature of the defect.[15]
Platelet disorders
Platelet disorders are either congenital or acquired. Examples of congenital platelet disorders are Glanzmann's thrombasthenia, Bernard–Soulier syndrome (abnormal glycoprotein Ib-IX-V complex), gray platelet syndrome (deficient alpha granules), and delta storage pool deficiency (deficient dense granules). Most are rare. They predispose to hemorrhage. Von Willebrand disease is due to deficiency or abnormal function of von Willebrand factor, and leads to a similar bleeding pattern; its milder forms are relatively common.
Decreased platelet numbers (thrombocytopenia) is due to insufficient production (e.g., myelodysplastic syndrome or other bone marrow disorders), destruction by the immune system (immune thrombocytopenic purpura/ITP), or consumption (e.g., thrombotic thrombocytopenic purpura/TTP, hemolytic-uremic syndrome/HUS, paroxysmal nocturnal hemoglobinuria/PNH, disseminated intravascular coagulation/DIC, heparin-induced thrombocytopenia/HIT). Most consumptive conditions lead to platelet activation, and some are associated with thrombosis.
Coagulation factor disorders
The best-known coagulation factor disorders are the hemophilias. The three main forms are hemophilia A (factor VIII deficiency), hemophilia B (factor IX deficiency or "Christmas disease") and hemophilia C (factor XI deficiency, mild bleeding tendency). Hemophilia A and B are X-linked recessive disorders, whereas Hemophilia C is a much more rare autosomal recessive disorder most commonly seen in Ashkenazi Jews.
Von Willebrand disease (which behaves more like a platelet disorder except in severe cases), is the most common hereditary bleeding disorder and is characterized as being inherited autosomal recessive or dominant. In this disease, there is a defect in von Willebrand factor (vWF), which mediates the binding of glycoprotein Ib (GPIb) to collagen. This binding helps mediate the activation of platelets and formation of primary hemostasis.
Bernard–Soulier syndrome is a defect or deficiency in GPIb. GPIb, the receptor for vWF, can be defective and lead to lack of primary clot formation (primary hemostasis) and increased bleeding tendency. This is an autosomal recessive inherited disorder.
Thrombasthenia of Glanzmann and Naegeli (Glanzmann thrombasthenia) is extremely rare. It is characterized by a defect in GPIIb/IIIa fibrinogen receptor complex. When GPIIb/IIIa receptor is dysfunctional, fibrinogen cannot cross-link platelets, which inhibits primary hemostasis. This is an autosomal recessive inherited disorder.
In liver failure (acute and chronic forms), there is insufficient production of coagulation factors by the liver; this may increase bleeding risk.
Deficiency of Vitamin K may also contribute to bleeding disorders because clotting factor maturation depends on Vitamin K.
Thrombosis is the pathological development of blood clots. These clots may break free and become mobile, forming an embolus or grow to such a size that occludes the vessel in which it developed. An embolism is said to occur when the thrombus (blood clot) becomes a mobile embolus and migrates to another part of the body, interfering with blood circulation and hence impairing organ function downstream of the occlusion. This causes ischemia and often leads to ischemic necrosis of tissue. Most cases of venous thrombosis are due to acquired states (older age, surgery, cancer, immobility) or inherited thrombophilias (e.g., antiphospholipid syndrome, factor V Leiden, and various other genetic deficiencies or variants).
Mutations in factor XII have been associated with an asymptomatic prolongation in the clotting time and possibly a tendency toward thrombophlebitis. Other mutations have been linked with a rare form of hereditary angioedema (type III) essentialism.
Pharmacology
Procoagulants
The use of adsorbent chemicals, such as zeolites, and other hemostatic agents are also used for sealing severe injuries quickly (such as in traumatic bleeding secondary to gunshot wounds). Thrombin and fibrin glue are used surgically to treat bleeding and to thrombose aneurysms.
Desmopressin is used to improve platelet function by activating arginine vasopressin receptor 1A.
Coagulation factor concentrates are used to treat hemophilia, to reverse the effects of anticoagulants, and to treat bleeding in patients with impaired coagulation factor synthesis or increased consumption. Prothrombin complex concentrate, cryoprecipitate and fresh frozen plasma are commonly used coagulation factor products. Recombinant activated human factor VII is increasingly popular in the treatment of major bleeding.
Tranexamic acid and aminocaproic acid inhibit fibrinolysis and lead to a de facto reduced bleeding rate. Before its withdrawal, aprotinin was used in some forms of major surgery to decrease bleeding risk and need for blood products.
Anticoagulants
Anticoagulants and anti-platelet agents are amongst the most commonly used medications. Anti-platelet agents include aspirin, dipyridamole, ticlopidine, clopidogrel, ticagrelor and prasugrel; the parenteral glycoprotein IIb/IIIa inhibitors are used during angioplasty. Of the anticoagulants, warfarin (and related coumarins) and heparin are the most commonly used. Warfarin affects the vitamin K-dependent clotting factors (II, VII, IX, X) and protein C and protein S, whereas heparin and related compounds increase the action of antithrombin on thrombin and factor Xa. A newer class of drugs, the direct thrombin inhibitors, is under development; some members are already in clinical use (such as lepirudin). Also in clinical use are other small molecular compounds that interfere directly with the enzymatic action of particular coagulation factors (the directly acting oral anticoagulants: dabigatran, rivaroxaban, apixaban, and edoxaban).[16]
Coagulation factors
| Number and/or name | Function | Associated genetic disorders |
|---|---|---|
| I (fibrinogen) | Forms clot (fibrin) | Congenital afibrinogenemia, Familial renal amyloidosis |
| II (prothrombin) | Its active form (IIa) activates I, V, VII, VIII, XI, XIII, protein C, platelets | Prothrombin G20210A, Thrombophilia |
| III (tissue factor or tissue thromboplastin) | Co-factor of VIIa (formerly known as factor III) | |
| IV (calcium) | Required for coagulation factors to bind to phospholipid (formerly known as factor IV) | |
| V (proaccelerin, labile factor) | Co-factor of X with which it forms the prothrombinase complex | Activated protein C resistance |
| VI | Unassigned – old name of Factor Va | |
| VII (stable factor, proconvertin) | Activates IX, X | congenital factor VII deficiency |
| VIII (Antihemophilic factor A) | Co-factor of IX with which it forms the tenase complex | Haemophilia A |
| IX (Antihemophilic factor B or Christmas factor) | Activates X: forms tenase complex with factor VIII | Haemophilia B |
| X (Stuart-Prower factor) | Activates II: forms prothrombinase complex with factor V | Congenital Factor X deficiency |
| XI (plasma thromboplastin antecedent) | Activates IX | Haemophilia C |
| XII (Hageman factor) | Activates factor XI, VII, prekallikrein and plasminogen | Hereditary angioedema type III |
| XIII (fibrin-stabilizing factor) | Crosslinks fibrin | Congenital Factor XIIIa/b deficiency |
| von Willebrand factor | Binds to VIII, mediates platelet adhesion | von Willebrand disease |
| prekallikrein (Fletcher factor) | Activates XII and prekallikrein; cleaves HMWK | Prekallikrein/Fletcher Factor deficiency |
| high-molecular-weight kininogen (HMWK) (Fitzgerald factor) | Supports reciprocal activation of XII, XI, and prekallikrein | Kininogen deficiency |
| fibronectin | Mediates cell adhesion | Glomerulopathy with fibronectin deposits |
| antithrombin III | Inhibits IIa, Xa, and other proteases | Antithrombin III deficiency |
| heparin cofactor II | Inhibits IIa, cofactor for heparin and dermatan sulfate ("minor antithrombin") | Heparin cofactor II deficiency |
| protein C | Inactivates Va and VIIIa | Protein C deficiency |
| protein S | Cofactor for activated protein C (APC, inactive when bound to C4b-binding protein) | Protein S deficiency |
| protein Z | Mediates thrombin adhesion to phospholipids and stimulates degradation of factor X by ZPI | Protein Z deficiency |
| Protein Z-related protease inhibitor (ZPI) | Degrades factors X (in presence of protein Z) and XI (independently) | |
| plasminogen | Converts to plasmin, lyses fibrin and other proteins | Plasminogen deficiency, type I (ligneous conjunctivitis) |
| alpha 2-antiplasmin | Inhibits plasmin | Antiplasmin deficiency |
| tissue plasminogen activator (tPA) | Activates plasminogen | Familial hyperfibrinolysis and thrombophilia |
| urokinase | Activates plasminogen | Quebec platelet disorder |
| plasminogen activator inhibitor-1 (PAI1) | Inactivates tPA & urokinase (endothelial PAI) | Plasminogen activator inhibitor-1 deficiency |
| plasminogen activator inhibitor-2 (PAI2) | Inactivates tPA & urokinase (placental PAI) | |
| cancer procoagulant | Pathological factor X activator linked to thrombosis in cancer |
History
Initial discoveries
Theories on the coagulation of blood have existed since antiquity. Physiologist Johannes Müller (1801–1858) described fibrin, the substance of a thrombus. Its soluble precursor, fibrinogen, was thus named by Rudolf Virchow (1821–1902), and isolated chemically by Prosper Sylvain Denis (1799–1863). Alexander Schmidt suggested that the conversion from fibrinogen to fibrin is the result of an enzymatic process, and labeled the hypothetical enzyme "thrombin" and its precursor "prothrombin".[17][18] Arthus discovered in 1890 that calcium was essential in coagulation.[19][20] Platelets were identified in 1865, and their function was elucidated by Giulio Bizzozero in 1882.[21]
The theory that thrombin is generated by the presence of tissue factor was consolidated by Paul Morawitz in 1905.[22] At this stage, it was known that thrombokinase/thromboplastin (factor III) is released by damaged tissues, reacting with prothrombin (II), which, together with calcium (IV), forms thrombin, which converts fibrinogen into fibrin (I).[23]
Coagulation factors
The remainder of the biochemical factors in the process of coagulation were largely discovered in the 20th century.
A first clue as to the actual complexity of the system of coagulation was the discovery of proaccelerin (initially and later called Factor V) by Paul Owren (1905–1990) in 1947. He also postulated its function to be the generation of accelerin (Factor VI), which later turned out to be the activated form of V (or Va); hence, VI is not now in active use.[23]
Factor VII (also known as serum prothrombin conversion accelerator or proconvertin, precipitated by barium sulfate) was discovered in a young female patient in 1949 and 1951 by different groups.
Factor VIII turned out to be deficient in the clinically recognised but etiologically elusive hemophilia A; it was identified in the 1950s and is alternatively called antihemophilic globulin due to its capability to correct hemophilia A.[23]
Factor IX was discovered in 1952 in a young patient with hemophilia B named Stephen Christmas (1947–1993). His deficiency was described by Dr. Rosemary Biggs and Professor R.G. MacFarlane in Oxford, UK. The factor is, hence, called Christmas Factor. Christmas lived in Canada, and campaigned for blood transfusion safety until succumbing to transfusion-related AIDS at age 46. An alternative name for the factor is plasma thromboplastin component, given by an independent group in California.[23]
Hageman factor, now known as factor XII, was identified in 1955 in an asymptomatic patient with a prolonged bleeding time named of John Hageman. Factor X, or Stuart-Prower factor, followed, in 1956. This protein was identified in a Ms. Audrey Prower of London, who had a lifelong bleeding tendency. In 1957, an American group identified the same factor in a Mr. Rufus Stuart. Factors XI and XIII were identified in 1953 and 1961, respectively.[23]
The view that the coagulation process is a "cascade" or "waterfall" was enunciated almost simultaneously by MacFarlane[24] in the UK and by Davie and Ratnoff[25] in the USA, respectively.
Nomenclature
The usage of Roman numerals rather than eponyms or systematic names was agreed upon during annual conferences (starting in 1955) of hemostasis experts. In 1962, consensus was achieved on the numbering of factors I–XII.[26] This committee evolved into the present-day International Committee on Thrombosis and Hemostasis (ICTH). Assignment of numerals ceased in 1963 after the naming of Factor XIII. The names Fletcher Factor and Fitzgerald Factor were given to further coagulation-related proteins, namely prekallikrein and high-molecular-weight kininogen, respectively.[23]
Factors III and VI are unassigned, as thromboplastin was never identified, and actually turned out to consist of ten further factors, and accelerin was found to be activated Factor V.
Other species
All mammals have an extremely closely related blood coagulation process, using a combined cellular and serine protease process. In fact, it is possible for any mammalian coagulation factor to "cleave" its equivalent target in any other mammal. The only non-mammalian animal known to use serine proteases for blood coagulation is the horseshoe crab.[27]
See also
References
- Furie B, Furie BC (2005). "Thrombus formation in vivo". J. Clin. Invest. 115 (12): 3355–62. doi:10.1172/JCI26987. PMC 1297262. PMID 16322780.
- David Lillicrap; Nigel Key; Michael Makris; Denise O'Shaughnessy (2009). Practical Hemostasis and Thrombosis. Wiley-Blackwell. pp. 1–5. ISBN 978-1-4051-8460-1.
- Alan D. Michelson (26 October 2006). Platelets. Academic Press. pp. 3–5. ISBN 978-0-12-369367-9. Retrieved 18 October 2012.
- Schmaier, Alvin H.; Lazarus, Hillard M. (2011). Concise guide to hematology. Chichester, West Sussex, UK: Wiley-Blackwell. p. 91. ISBN 978-1-4051-9666-6.
- Nigel Key; Michael Makris; et al. (2009). Practical Hemostasis and Thrombosis. Wiley-Blackwell. pp. 2. ISBN 978-1-4051-8460-1.
- Pallister CJ, Watson MS (2010). Haematology. Scion Publishing. pp. 334–336. ISBN 978-1-904842-39-2.
- Pallister CJ, Watson MS (2010). Haematology. Scion Publishing. pp. 336–347. ISBN 978-1-904842-39-2.
- "COAGULATION FACTOR". Clotbase.bicnirrh.res.in. Retrieved 20 May 2018.
- Hoffbrand, A. V. (2002). Essential haematology. Oxford: Blackwell Science. pp. 241–243. ISBN 978-0-632-05153-3.
- Long, Andrew T.; Kenne, Ellinor; Jung, Roman; Fuchs, Tobias A.; Renné, Thomas (2015). "Contact system revisited: An interface between inflammation, coagulation, and innate immunity". Journal of Thrombosis and Haemostasis. 14 (3): 427–437. doi:10.1111/jth.13235. PMID 26707513.
- Hoffman, M (August 2003). "Remodeling the blood coagulation cascade". Journal of Thrombosis and Thrombolysis. 16 (1–2): 17–20. doi:10.1023/B:THRO.0000014588.95061.28. ISSN 1573-742X. PMID 14760207.
- Hoffbrand, A. V. (2002). Essential haematology. Oxford: Blackwell Science. pp. 243–245. ISBN 978-0-632-05153-3.
- Immunology – Chapter One: Innate ot non-specific immunity Gene Mayer, Ph.D. Immunology Section of Microbiology and Immunology On-line. University of South Carolina
- David Lillicrap; Nigel Key; Michael Makris; Denise O'Shaughnessy (2009). Practical Hemostasis and Thrombosis. Wiley-Blackwell. pp. 7–16. ISBN 978-1-4051-8460-1.
- Hatton, Chris (2008). Haematology (Lecture Notes). Cambridge, MA: Blackwell Publishers. pp. 145–166. ISBN 978-1-4051-8050-4.
- Soff GA (March 2012). "A new generation of oral direct anticoagulants". Arteriosclerosis, Thrombosis, and Vascular Biology. 32 (3): 569–74. doi:10.1161/ATVBAHA.111.242834. PMID 22345595.
- Schmidt A (1872). "Neue Untersuchungen über die Faserstoffgerinnung". Pflügers Archiv für die gesamte Physiologie. 6: 413–538. doi:10.1007/BF01612263.
- Schmidt A. Zur Blutlehre. Leipzig: Vogel, 1892.
- Arthus M, Pagès C (1890). "Nouvelle theorie chimique de la coagulation du sang". Arch Physiol Norm Pathol. 5: 739–46.
- Shapiro SS (2003). "Treating thrombosis in the 21st century". N. Engl. J. Med. 349 (18): 1762–4. doi:10.1056/NEJMe038152. PMID 14585945.
- Brewer DB (2006). "Max Schultze (1865), G. Bizzozero (1882) and the discovery of the platelet". Br. J. Haematol. 133 (3): 251–8. doi:10.1111/j.1365-2141.2006.06036.x. PMID 16643426.
- Morawitz P (1905). "Die Chemie der Blutgerinnung". Ergebn Physiol. 4: 307–422. doi:10.1007/BF02321003.
- Giangrande PL (2003). "Six characters in search of an author: the history of the nomenclature of coagulation factors". Br. J. Haematol. 121 (5): 703–12. doi:10.1046/j.1365-2141.2003.04333.x. PMID 12780784.
- MacFarlane RG (1964). "An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier". Nature. 202 (4931): 498–9. Bibcode:1964Natur.202..498M. doi:10.1038/202498a0. PMID 14167839.
- Davie EW, Ratnoff OD (1964). "Waterfall sequence for intrinsic blood clotting". Science. 145 (3638): 1310–2. Bibcode:1964Sci...145.1310D. doi:10.1126/science.145.3638.1310. PMID 14173416.
- Wright IS (1962). "The Nomenclature of Blood Clotting Factors". Can Med Assoc J. 86 (8): 373–4. PMC 1848865. PMID 14008442.
- Osaki T, Kawabata S (June 2004). "Structure and function of coagulogen, a clottable protein in horseshoe crabs". Cellular and Molecular Life Sciences. 61 (11): 1257–65. doi:10.1007/s00018-004-3396-5. PMID 15170505.
Further reading
- Hoffman, M; Monroe DM, 3rd (June 2001). "A cell-based model of hemostasis". Thrombosis and Haemostasis. 85 (6): 958–65. doi:10.1055/s-0037-1615947. PMID 11434702.
- Hoffman, Maureane; Monroe, Dougald M. (2007). "Coagulation 2006: A Modern View of Hemostasis". Hematology/Oncology Clinics of North America. 21 (1): 1–11. doi:10.1016/j.hoc.2006.11.004. PMID 17258114.