Glycoprotein Ib-IX-V Receptor Complex
The GPIb-IX-V complex is a profuse membrane receptor complex originating in megakaryocytes and exclusively functional on the surface of platelets.[1] It primarily functions to mediate the first critical step in platelet adhesion, by facilitating binding to von Willebrand factor (VWF) on damaged sub-endothelium under conditions of high fluid shear stress.[2][3] Although the primary ligand for the GPIb-V-IX receptor is VWF, it can also bind to a number of other ligands in the circulation such as thrombin, P-selectin, factor XI, factor XII, high molecular weight kininogen as well as bacteria. GPIb-IX-V offers a critical role in thrombosis, metastasis, and the life cycle of platelets, and is implicated in a number of thrombotic pathological processes such as stroke or myocardial infarction.[1][2]
Molecular structure
Overview
GPIb-IX-V consists of four different subunits namely: GPIbα (molecular weight (MW) 135 kDa), GPIbβ (MW 26 kDa), GPIX (MW 20 kDa) and GPV (MW 82kDa). The complex is assembled such that GPIbα, GPIbβ and GPIX form a highly integrated protein complex in a 1:2:1 stoichiometry; and this associates weakly with GPV resulting in an overall stoichiometric ratio of 1:1.[1][4][5][6]
Each subunit of the complex is a type I transmembrane (TM) protein which consists of a leucine-rich repeat (LRR) ectodomain (extracellular domain), a single transmembrane helix, and a relatively short cytoplasmic tail that lacks enzymatic activity.[1][7]
The quaternary stabilization of the receptor is facilitated by covalent and non-covalent interactions. The GPIbα subunit is linked to two GPIbβ subunits via membrane-proximal disulfide bonds, while GPIX associates itself tightly through non-covalent interactions with GPIb.[4][5][7] The concomitant expression of all three subunits is required to allow the effective expression of GPIb-IX on the platelet cell surface and analysis of receptor expression in transfected Chinese hamster ovary (CHO) cells has further supported that the interaction between these subunits also acts to stabilize them.[1]
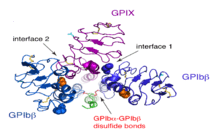
Each of the four subunits (GPIbα, GPIbβ, GPIX and GPV) is part of the leucine rich repeat motif superfamily. These leucine rich repeat sequences tend to be about 24 amino acids in length either occurring singly or in tandem repeats flanked by conserved N-terminal and C-terminal disulfide loop structures.[3] Nevertheless, even though these structural similarities exist, distinctive genes that exist on different chromosomes of the genome code for the polypeptides that make up the GPIb-V-IX complex.
The four genes that code for the components of the receptor in humans have a simple organization in which the coding sequence is contained within a single exon. This is with the exception of the gene for GPIbβ, which contains an intron 10 bases following the start codon.[3]
Human GPIbα is the product of a gene on chromosome 17 specifically 17p12, GPIbβ is the product of a gene on chromosome 22 specifically 22q11.2, while GPV and GPIX are products of genes found on chromosome 3 specifically 3q21 and 3q29 respectively.[8] Under normal conditions, all four molecules are expressed exclusively in the platelet lineage. GPIbα, GPIbβ and GPIX are necessary for the effective biosynthesis of the receptor and are closely associated at the platelet membrane. Typically, a lack of a single subunit significantly decreases the surface expression of the entire receptor complex.[8][9]
GPIbα
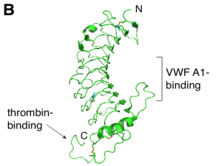
GPIbα (CD42b) consisting of 610 amino acids is the major subunit and contains all known extracellular ligand-binding sites of the complex for example: the A1 domain of von Willebrand factor (VWF) has a binding region as marked in the N-terminal domain of GPIbα; while the thrombin binding site is contained in a conformationally flexible acidic residue-rich sequence containing sulfated tyrosines.[1][3]
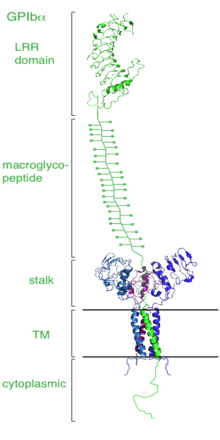
Dissection of the crystal structure of the GPIbα N-terminal leucine rich repeat domain discloses the presence of a single disulfide bond between cysteine (Cys) residues Cys4 and Cys17 in the N-capping region, and two disulfide bonds (Cys209-Cys248 and Cys211-Cys264) in the C-capping region. Furthermore, there are seven tandem leucine rich repeats and their flanking sequences in the central parallel β-coil region. This parallel β-coil region is made up of three sided coils stacked in layers and contains two asparagine residues (Asn21 and Asn159), which serve as N-glycosylation sites. Following the leucine rich repeat domain is the acidic residue-rich sequence containing sulfated tyrosines, the highly O-glycosylated macroglycopeptide, a stalk region of about 40 to 50 residues, a single transmembrane sequence and finally a cytoplasmic tail containing 96 amino acid residues which includes serine residues such as Ser587, Ser590 and Ser609 that can be phosphorylated.[1][3]
GPIbβ, GPIX, GPV
GPIbβ (CD42c) contains 181 amino acids. In the extracellular domain (ectodomain), both the N-capping and C-capping regions, which flank the leucine rich repeat sequence, contain two interlocking disulfide bonds. Furthermore, there is only a single leucine-rich repeat giving rise to a much less curved parallel β-coil region as compared to that in GPIbα. GPIbβ contains only one N-glycosylation site (Asn41) and is disulfide linked to GPIbα immediately proximal to the plasma membrane of the platelet via Cys122 located at the junction of the extracellular and transmembrane domains.[1][3]
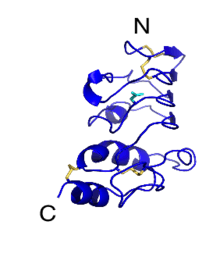
The GPIbβ cytoplasmic domain has a sequence of 34 amino acids. The region adjacent to the membrane is enriched in basic residues and Ser166 found more distally is phosphorylated and appears to have a role in platelet cytoskeletal rearrangement.
GPIX (CD42a) contains 160 amino acids. The extracellular domain, which also only has a single leucine rich repeat sequence shares more than 45% sequence identity with GPIbβ counterpart. However, the transmembrane and cytoplasmic sequences are considerably different. The GPIX cytoplasmic tail is short consisting of 8 residues and is not known to associate with intracellular proteins. There is also a cysteine residue (Cys154) located at the junction of the transmembrane and cytoplasmic domains. The extracellular domain of GPV contains 13 leucine rich repeats flanked by N- and C- capping regions both containing two interlocking disulfide bonds. This is followed by a stalk region, the transmembrane sequence and a short cytoplasmic tail rich in basic residues.[1][3]
The GPV (CD42d) subunit is only weakly associated with the GPIb-IX part of the receptor complex through interactions between the transmembrane domains and has little impact on the surface expression of GPIb-IX, although GPIb-IX is required for efficient expression of GPV.[1][6] Furthermore, GPV doesn’t appear to be critical for VWF binding or signal transduction.[7]
Role in disease
Abnormalities of the GPIb-V-IX complex result in abnormal appearance and functioning of platelets resulting in Bernard–Soulier syndrome (BSS), a condition first described by Bernard J and Soulier J.P.[10] It is a rare hereditary bleeding disorder most commonly with an autosomal recessive inheritance and diagnosed based on prolonged skin-bleeding time, a reduced number of very large platelets (macrothrombocytopenia) and defective ristocetin-induced platelet agglutination.[11]
Bernard Soulier Syndrome is characterized by little or no expression of GPIb-IX on the surface of platelets which in turn has the same effect on GPV. There have been a number of mutations associated with BSS patients that have been mapped to GPIbα, GPIbβ and GPIX demonstrating that all three subunits are required for effective surface expression of the complex on platelets.[7]
References
- Li R, Emsley J (April 2013). "The organizing principle of the platelet glycoprotein Ib-IX-V complex". J. Thromb. Haemost. 11 (4): 605–14. doi:10.1111/jth.12144. PMC 3696474. PMID 23336709.
- McEwan PA, Andrews RK, Emsley J (November 2009). "Glycoprotein Ibalpha inhibitor complex structure reveals a combined steric and allosteric mechanism of von Willebrand factor antagonism". Blood. 114 (23): 4883–5. doi:10.1182/blood-2009-05-224170. PMID 19726719.
- López JA, Andrews RK, Afshar-Kharghan V, Berndt MC (June 1998). "Bernard-Soulier syndrome". Blood. 91 (12): 4397–418. doi:10.1182/blood.V91.12.4397. PMID 9616133.
- Du X, Beutler L, Ruan C, Castaldi PA, Berndt MC (May 1987). "Glycoprotein Ib and glycoprotein IX are fully complexed in the intact platelet membrane". Blood. 69 (5): 1524–7. doi:10.1182/blood.V69.5.1524.1524. PMID 2436691.
- Luo SZ, Mo X, Afshar-Kharghan V, Srinivasan S, López JA, Li R (January 2007). "Glycoprotein Ibalpha forms disulfide bonds with 2 glycoprotein Ibbeta subunits in the resting platelet". Blood. 109 (2): 603–9. doi:10.1182/blood-2006-05-024091. PMC 1785083. PMID 17008541.
- Mo X, Liu L, López JA, Li R (September 2012). "Transmembrane domains are critical to the interaction between platelet glycoprotein V and glycoprotein Ib-IX complex". J. Thromb. Haemost. 10 (9): 1875–86. doi:10.1111/j.1538-7836.2012.04841.x. PMC 3499136. PMID 22759073.
- McEwan PA, Yang W, Carr KH, et al. (November 2011). "Quaternary organization of GPIb-IX complex and insights into Bernard–Soulier syndrome revealed by the structures of GPIbβ and a GPIbβ/GPIX chimera". Blood. 118 (19): 5292–301. doi:10.1182/blood-2011-05-356253. PMC 3217411. PMID 21908432.
- Lanza F (2006). "Bernard-Soulier syndrome (hemorrhagiparous thrombocytic dystrophy)". Orphanet J Rare Dis. 1: 46. doi:10.1186/1750-1172-1-46. PMC 1660532. PMID 17109744.
- Nurden AT (August 2005). "Qualitative disorders of platelets and megakaryocytes". J. Thromb. Haemost. 3 (8): 1773–82. doi:10.1111/j.1538-7836.2005.01428.x. PMID 16102044.
- Bernard J, Soulier JP (1948). "Sur une nouvelle variete de dystrophie thrombocythaire hemorragipare congenitale". Sem Hop Paris. 24: 3217–3223.
- Strassel C, David T, Eckly A, et al. (January 2006). "Synthesis of GPIb beta with novel transmembrane and cytoplasmic sequences in a Bernard–Soulier patient resulting in GPIb-defective signaling in CHO cells". J. Thromb. Haemost. 4 (1): 217–28. doi:10.1111/j.1538-7836.2005.01654.x. PMID 16409472.