Central core disease
Central core disease (CCD), also known as central core myopathy, is an autosomal dominantly inherited[1] muscle disorder present from birth that negatively affects the skeletal muscles. It was first described by Shy and Magee in 1956.[2][3] It is characterized by the appearance of the myofibril under the microscope.[4]
| Central Core Disease | |
|---|---|
| Other names | Central core myopathy |
.jpg) | |
| Histopathologic appearance of typical central core disease: NADH-TR, transverse section from the rectus femoris. Marked predominance of dark staining, high oxidative type 1 fibres with cores affecting the majority of fibres. Cores are typically well demarcated and centrally located (→), but may occasionally be multiple and of eccentric location. | |
| Specialty | Neurology |
Signs and symptoms
The symptoms of CCD are variable, but usually involve hypotonia (decreased muscle tone) at birth, mild delay in child development (highly variable between cases), weakness of the facial muscles, and skeletal malformations such as scoliosis and hip dislocation.[2]
CCD is usually diagnosed in infancy or childhood, but some patients remain asymptomatic until adulthood to middle age.[5] While generally not progressive, there appears to be a growing number of people who do experience a slow clinically significant progression of symptomatology. These cases may be due to the large number of mutations of ryanodine receptor malfunction, and with continued research may be found to be clinical variants.
Pathophysiology
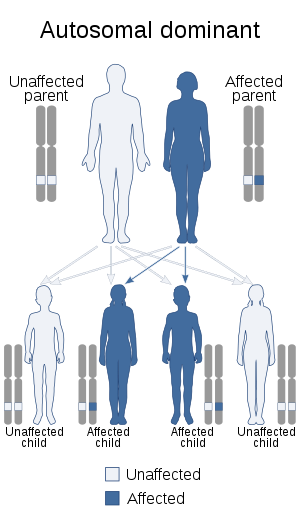
Central core disease is inherited in an autosomal dominant fashion. Most cases have demonstrable mutations in the ryanodine receptor type 1 (RYR1) gene,[1] which are often de novo (newly developed). People with CCD are at increased risk for developing malignant hyperthermia (MH) when receiving general anesthesia.[2]
Diagnosis
The diagnosis is made based on the combination of typical symptoms and the appearance on biopsy (tissue sample) from muscle. The name derives from the typical appearance of the biopsy on light microscopy, where the muscle cells have cores that are devoid of mitochondria and specific enzymes.[2]
Respiratory insufficiency develops in a small proportion of cases. Creatine kinase tend to be normal and electromyography (EMG) shows short duration, short amplitude motor unit action potentials.[2]
Treatment
There is no specific treatment but triggering anesthetics are avoided and relatives are screened for RYR1 mutations as these may make them susceptible to MH.[2]
References
- Robinson, Rl; Brooks, C; Brown, Sl; Ellis, Fr; Halsall, Pj; Quinnell, Rj; Shaw, Ma; Hopkins, Pm (August 2002). "RYR1 mutations causing central core disease are associated with more severe malignant hyperthermia in vitro contracture test phenotypes". Human Mutation. 20 (2): 88–97. doi:10.1002/humu.10098. PMID 12124989.
- Quinlivan RM, Muller CR, Davis M, et al. (2003). "Central core disease: clinical, pathological, and genetic features". Arch. Dis. Child. 88 (12): 1051–5. doi:10.1136/adc.88.12.1051. PMC 1719384. PMID 14670767.
- Magee KR, Shy GM (1956). "A new congenital non-progressive myopathy". Brain. 79 (4): 610–21. CiteSeerX 10.1.1.1026.496. doi:10.1093/brain/79.4.610. PMID 13396066.
- "central core disease" at Dorland's Medical Dictionary
- Talwalkar, SS; Parker, JR; Heffner, RR; Parker, JC (2006). "Adult central core disease. Clinical, histologic and genetic aspects: case report and review of the literature". Clin Neuropathol. 25 (4): 180–4. PMID 16866299.
External links
| Classification | |
|---|---|
| External resources |