Cancer syndrome
A cancer syndrome, or family cancer syndrome, is a genetic disorder in which inherited genetic mutations in one or more genes predispose the affected individuals to the development of cancers and may also cause the early onset of these cancers. Cancer syndromes often show not only a high lifetime risk of developing cancer, but also the development of multiple independent primary tumors.[1]
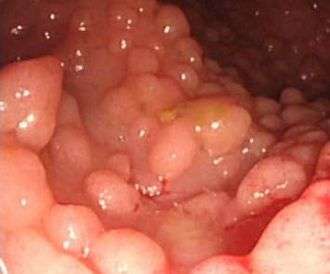
Many of these syndromes are caused by mutations in tumor suppressor genes, genes that are involved in protecting the cell from turning cancerous. Other genes that may be affected are DNA repair genes, oncogenes and genes involved in the production of blood vessels (angiogenesis).[2] Common examples of inherited cancer syndromes are hereditary breast-ovarian cancer syndrome and hereditary non-polyposis colon cancer (Lynch syndrome).[3][4]
Background
Hereditary cancer syndromes underlie 5 to 10% of all cancers and there are over 50 identifiable hereditary forms of cancer.[5] Scientific understanding of cancer susceptibility syndromes is actively expanding: additional syndromes are being found,[6] the underlying biology is becoming clearer, and commercialization of diagnostic genetics methodology is improving clinical access. Given the prevalence of breast and colon cancer, the most widely recognized syndromes include hereditary breast-ovarian cancer syndrome (HBOC) and hereditary non-polyposis colon cancer (HNPCC, Lynch syndrome).[6]
Some rare cancers are strongly associated with hereditary cancer predisposition syndromes. Genetic testing should be considered with adrenocortical carcinoma; carcinoid tumors; diffuse gastric cancer; fallopian tube/primary peritoneal cancer; leiomyosarcoma; medullary thyroid cancer; paraganglioma/pheochromocytoma; renal cell carcinoma of chromophobe, hybrid oncocytic, or oncocytoma histology; sebaceous carcinoma; and sex cord tumors with annular tubules.[6] Primary care physicians can identify people who are at risk of heridatary cancer syndrome.[7]
Genetics of cancer
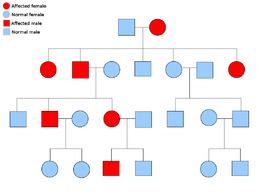
Two copies of every gene are present in all cells of the body and each one is called an allele. Most cancer syndromes are transmitted in a mendelian autosomal dominant manner. In these cases, only one faulty allele has to be present for an individual to have a predisposition to cancer. Individuals with one normal allele and one faulty allele are known as heterozygous. A heterozygous individual and a person with two normal alleles (homozygous) will have a 50% chance of producing an affected child.[8] The mutation in the inherited gene is known as a germline mutation and a further mutation in the normal allele results in the development of cancer. This is known as Knudson's two-hit hypothesis, where the first hit of the gene is the inherited mutation and the second hit occurs later in life.[2] As only one allele needs to be mutated (as compared to both in so-called "sporadic cancers"), the individual has a higher chance of developing the cancer than the general population.
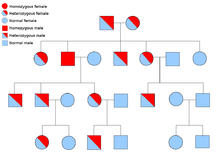
Less often, syndromes may be transmitted as an autosomal recessive trait. Both alleles of a gene must be mutated in autosomal recessive disorders for an individual to have a predisposition to cancer. A person with two recessive alleles is known as homozygous recessive. Both parents must have at least one faulty allele in order for a child to be homozygous recessive. If both parents have one mutant allele and one normal allele (heterozygous) then they have a 25% chance of producing a homozygous recessive child (has predisposition), 50% chance of producing a heterozygous child (carrier of the faulty gene) and 25% chance of produced a child with two normal alleles.[8]
Examples of autosomal dominant cancer syndromes are autoimmune lymphoproliferative syndrome (Canale-Smith syndrome), Beckwith–Wiedemann syndrome (although 85% of cases are sporadic), Birt–Hogg–Dubé syndrome, Carney syndrome, familial chordoma, Cowden syndrome, dysplastic nevus syndrome with familial melanoma, familial adenomatous polyposis, hereditary breast–ovarian cancer syndrome, hereditary diffuse gastric cancer (HDGC), Hereditary nonpolyposis colorectal cancer (Lynch syndrome), Howel–Evans syndrome of eosophageal cancer with tylosis, juvenile polyposis syndrome, Li–Fraumeni syndrome, multiple endocrine neoplasia type 1/2, multiple osteochondromatosis, neurofibromatosis type 1/2, nevoid basal-cell carcinoma syndrome (Gorlin syndrome), Peutz–Jeghers syndrome, familial prostate cancer, hereditary leiomyomatosis renal cell cancer (LRCC), hereditary papillary renal cell cancer (HPRCC), hereditary paraganglioma-pheochromocytoma syndrome, retinoblastoma, tuberous sclerosis, von Hippel–Lindau disease and Wilm's tumor.[9]
Examples of autosomal recessive cancer syndromes are ataxia–telangiectasia, Bloom syndrome, Fanconi anemia, MUTYH-associated polyposis, Rothmund–Thomson syndrome, Werner syndrome and Xeroderma pigmentosum.[9]
Examples
Although cancer syndromes exhibit an increased risk of cancer, the risk varies. For some of these diseases, cancer is not their primary feature. The discussion here focuses on their association with an increased risk of cancer. This list is far from exhaustive.
Fanconi anemia
Fanconi anemia (FA) is a disorder with a wide clinical spectrum, including: early onset and increased risk of cancer; bone marrow failure; and congenital abnormalities. The most prominent manifestations of this disorder are those related to hematopoeisis (production of blood by the bone marrow); these include aplastic anemia, myelodysplastic syndrome and acute myeloid leukemia. Hepatic tumors and squamous cell carcinomas of the esophagus, oropharynx and uvula are solid tumors commonly linked to FA. Congenital abnormalities include: skeletal anomalies (especially those affecting the hands), cafe au lait spots and hypopigmentation. To date, the genes known to cause FA are: FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ, FANCL, FANCM, FANCN, FANCO, FANCP and BRCA2 (previously known as FANCD1). Inheritance of this syndrome is primarily autosomal recessive, but FANCB can be inherited from the maternal or paternal x-chromosome (x-linked recessive inheritance). The FA pathway is involved in DNA repair when the two strands of DNA are incorrectly joined together (interstrand crosslinks). Many pathways are coordinated by the FA pathway for this including nucleotide excision repair, translesion synthesis and homologous recombination.[10][11][12][13][14]
Familial adenomatous polyposis
Familial adenomatous polyposis (FAP) is an autosomal dominant syndrome that greatly increases the risk of colorectal cancer. Around 1 in 8000 people will have this disease and it has approximately 100% penetrance. An individual with this disease will have hundreds to thousands of benign adenomas throughout their colon, which will in most cases progress to cancer. Other tumors increased in frequency include; osteomas, adrenal adenomas and carcinomas, thyroid tumors and desmoid tumors. The cause of this disorder is a mutated APC gene, which is involved in β-catenin regulation. Faulty APC causes β-catenin to accumulate in cells and activate transcription factors involved in cell proliferation, migration, differentiation and apoptosis (programmed cell death).[15][16][17]
Hereditary breast and ovarian cancer
Hereditary breast-ovarian cancer syndrome (HBOC) is an autosomal dominant genetic disorder caused by genetic mutations of the BRCA1 and BRCA2 genes. In women this disorder primarily increases the risk of breast and ovarian cancer, but also increases the risk of fallopian tube carcinoma and papillary serous carcinoma of the peritoneum. In men the risk of prostate cancer is increased. Other cancers that are inconsistently linked to this syndrome are pancreatic cancer, male breast cancer, colorectal cancer and cancers of the uterus and cervix. Genetic mutations account for approximately 7% and 14% of breast and ovarian cancer, respectively, and BRCA1 and BRCA2 account for 80% of these cases. BRCA1 and BRCA2 are both tumor suppressor genes implicated in maintaining and repairing DNA, which in turn leads to genome instability. Mutations in these genes allow further damage to DNA, which can lead to cancer.[18][19]
Hereditary non-polyposis colon cancer
Hereditary non-polyposis colon cancer (HNPCC), also known as Lynch syndrome, is an autosomal dominant cancer syndrome that increases the risk of colorectal cancer. It is caused by genetic mutations in DNA mismatch repair (MMR) genes, notably MLH1, MSH2, MSH6 and PMS2. In addition to colorectal cancer many other cancers are increased in frequency. These include; endometrial cancer, stomach cancer, ovarian cancer, cancers of the small bowel and pancreatic cancer. HNPCC is also associated with an early onset of colorectal cancer. MMR genes are involved in repairing DNA when the bases on each strand of DNA do not match. Defective MMR genes allow continuous insertion and deletion mutations in regions of DNA known as microsatellites. These short repetitive sequences of DNA become unstable, leading to a state of microsatellite instability (MSI). Mutated microsatellites are often found in genes involved in tumor initiation and progression, and MSI can enhance the survival of cells, leading to cancer.[4][20][21][22]
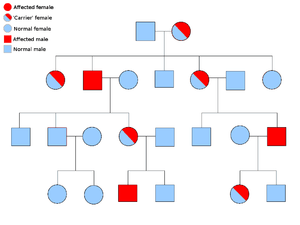
Hereditary paraganglioma-pheochromocytoma syndrome
Most cases of familial paraganglioma are caused by mutations in the succinate dehydrogenase (SDH; succinate:ubiquinone oxidoreductase) subunit genes (SDHD, SDHAF2, SDHC, SDHB).
PGL-1 is associated with SDHD mutation, and most PGL-1 individuals with paraganglioma have affected fathers rather than affected mothers. PGL1 and PGL2 are autosomal dominant with imprinting. PGL-4 is associated with SDHB mutation and is associated with a higher risk of pheochromocytoma, as well as renal cell cancer and non-medullary thyroid cancer.[23]
Li-Fraumeni syndrome
Li-Fraumeni syndrome is an autosomal dominant syndrome primarily caused by mutations in the TP53 gene, which greatly increases the risk of many cancers and is also highly associated with early onset of these cancers. Cancers linked to this disorder include; soft tissue sarcomas (often found in childhood), osteosarcoma, breast cancer, brain cancer, leukaemia and adrenocortical carcinoma. Individuals with Li-Fraumeni syndrome often have multiple independent primary cancers. The reason for the large clinical spectrum of this disorder may be due to other gene mutations that modify the disease. The protein produced by the TP53 gene, p53, is involved in cell cycle arrest, DNA repair and apoptosis. Defective p53 may not be able to properly perform these processes, which may be the reason for tumor formation. Because only 60-80% of individuals with the disorder have detectable mutations in TP53, other mutations in the p53 pathway may be involved in Li-Fraumeni syndrome.[24][25][26][27]
MUTYH-associated polyposis
MUTYH-associated polyposis shares most of its clinical features with FAP; the difference is that it is an autosomal recessive disorder caused by mutations in the MUTYH DNA repair gene. Tumors with increased risk in this disorder are colorectal cancer, gastric adenomas and duodenal adenomas.[15][28]
Nevoid basal cell carcinoma syndrome
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome, is an autosomal dominant cancer syndrome in which the risk of basal cell carcinoma is very high. The disease is characterized by basal cell nevi, jaw keratocysts and skeletal abnormalities. Estimates of NBCCS prevalence varies, but is approximately 1 in 60000. The presence of basal cell carcinoma is much higher in white than black individuals; 80% and 38%, respectively. Odontogenic keratocysts are found in approximately 75% of individuals with the disease and often occur early in life. The most common skeletal abnormalities occur in the head and face, but other areas are often affected such as the rib cage. The causative genetic mutation of this disease occurs in the PTCH gene, and the product of PTCH is a tumor suppressor involved in cell signaling. Although the exact role of this protein in NBCCS is not known, it is involved in the hedgehog signaling pathway, known to control cell growth and development.[29][30]
Von Hippel–Lindau disease
Von Hippel–Lindau (VHL) disease is a rare, autosomal dominant genetic condition that predisposes individuals to benign and malignant tumors. The most common tumors in VHL are central nervous system and retinal hemangioblastomas, clear cell renal carcinomas, pheochromocytomas, pancreatic neuroendocrine tumours, pancreatic cysts, endolymphatic sac tumors and epididymal papillary cystadenomas.[31][32] VHL results from a mutation in the von Hippel–Lindau tumor suppressor gene on chromosome 3p25.3.[33]
Xeroderma pigmentosum
Xeroderma pigmentosum (XP) is an autosomal recessive disorder characterized by sensitivity to ultra-violet (UV) light, massively increased risk of sunburn and increased risk of skin cancers. The risk of skin cancer is more than 10000 times that of normal individuals and includes many types of skin cancer, including melanoma and non-melanoma skin cancers. Also, sun exposed areas of the tongue, lips and eyes have an increased risk of becoming cancerous. XP may be associated with other internal cancers and benign tumors. In addition to cancer, some genetic mutations that cause XP are associated with neurodegeneration. XP may be caused by genetic mutations in 8 genes, which produce the following enzymes: XPA, XPB, XPC, XPD, XPE, XPF, XPG and Pol η. XPA-XPF are nucleotide excision repair enzymes that repair UV light-damaged DNA and faulty proteins will allow the buildup of mutations caused by UV light. Pol η is a polymerase, which is an enzyme involved in DNA replication. There are many polymerases, but pol η is the enzyme that replicates UV light-damaged DNA. Mutations in this gene will produce a faulty pol η enzyme that cannot replicate DNA with UV light damage. Individuals with mutations of this gene have a subset of XP; XP-variant disease.[34][35]
DNA repair defects and increased cancer risk
Many cancer syndromes are due to an inherited impairment in DNA repair capability. When an inherited mutation is present in a DNA repair gene, the repair gene will either not be expressed or expressed in an altered form. Then the repair function will likely be deficient, and, as a consequence, DNA damages will tend to accumulate. Such DNA damages can cause errors during DNA synthesis leading to mutations, some of which may give rise to cancer. Germ-line DNA repair mutations that increase the risk of cancer are listed in the Table.
| DNA repair gene | Protein | Repair pathways affected* | Cancers with increased risk |
|---|---|---|---|
| ataxia telangiectasia mutated | ATM | Different mutations in ATM reduce HRR, SSA or NHEJ [36] | leukemia, lymphoma, breast [36][37] |
| Bloom syndrome | BLM (helicase) | HRR [38] | leukemia, lymphoma, colon, breast, skin, lung, auditory canal, tongue, esophagus, stomach, tonsil, larynx, uterus [39] |
| breast cancer 1 & 2 | BRCA1 BRCA2 | HRR of double strand breaks and daughter strand gaps[40] | breast, ovarian [41] |
| Fanconi anemia genes FANCA,B,C,D1,D2,E,F,G,I,J,L,M,N,O,P | FANCA etc. | HRR and TLS [42] | leukemia, liver tumors, solid tumors many areas [43] |
| Hereditary nonpolyposis colorectal cancer genes MSH2 MSH6 MLH1 PMS2 | MSH2 MSH6 MLH1 PMS2 | MMR [44] | colorectal, endometrial, ovariain, gastrointestinal tract (stomach and small intestine, pancreas, biliary tract), urinary tract, brain (glioblastomas), and skin (keratoacanthomas and
sebaceous adenomas) [45] |
| Li-Fraumeni syndrome gene TP53 | P53 | Direct role in HRR, BER, NER and acts in DNA damage response[46] for those pathways and for NHEJ and MMR [47] | sarcomas, breast cancers, brain tumors, and adrenocortical carcinomas [48] |
| MRE11A | MRE11 | HRR and NHEJ [49] | breast [50] |
| MUTYH | MUTYH glycosylase | BER of A paired with 8-oxo-dG [51] | colorectal, duodenal, ovarian, bladder and skin cancers [52] |
| Nijmegen breakage syndrome | NBS (NBN) | NHEJ [53] | lymphoid cancers [53] |
| NTHL1 | NTHL1 | BER for Tg, FapyG, 5-hC, 5-hU in dsDNA[54] | Colon cancer, endometrial cancer, duodenal cancer, basal-cell carcinoma[55] |
| RECQL4 | RECQ4 | Helicase likely active in HRR [56] | basal cell carcinoma, squamous cell carcinoma, intraepidermal carcinoma [57] |
| Werner syndrome gene WRN | Werner syndrome ATP-dependent helicase | HRR, NHEJ, long patch BER [58] | soft tissue sarcoma, colorectal, skin, thyroid, pancreas [59] |
| Xeroderma pigmentosum genes XPA, XPB, XPD, XPF, XPG | XPA XPB XPD XPF XPG | Transcription coupled NER repairs the transcribed strands of transcriptionally active genes [60] | skin cancer (melanoma and non-melanoma) [60] |
| Xeroderma pigmentosum genes XPC, XPE (DDB2) | XPC, XPE | Global genomic NER, repairs damage in both transcribed and untranscribed DNA [61][62] | skin cancer (melanoma and non-melanoma) [61][62] |
| XPV (also called polymerase H) | DNA polymerase eta (Pol η) | Translesion synthesis (TLS) [63] | skin cancers (basal cell, squamous cell, melanoma) [63] |
- The acronyms for DNA repair pathways are HRR homologous recombinational repair, SSA sub-pathway of HRR, NHEJ non-homologous end joining, BER base excision repair, TLS translesion synthesis, NER nucleotide excision repair, MMR mismatch repair.
Genetic screening
Genetic testing can be used to identify mutated genes or chromosomes that are passed through generations. People who test positive for having a genetic mutation are not necessarily condemned to develop the cancer linked with the mutation, however they possess an increased risk of developing cancer in comparison to the general population. It is advised that people get a genetic test if their family medical history includes: Multiple family members with cancer, someone in their family that got cancer at a particularly young age or by being part of a certain ethnic group.[64]
The process of genetic screening is a simple, non-invasive procedure. However, before genes are tested for mutations the patient usually must go to a health care provider and go through a one-on-one consultation, where they discuss both the personal and family history of cancer. The medical professional can then assess the likelihood of the patient having the mutation and can guide them through the process that is genetic screening.[65] It is important that this consultation takes place because it ensures that the person gives informed consent to engage in genetic testing, is aware and understands the steps, benefits and limitations of the procedure and is more knowledgeable of the consequences of hearing test results.[66] The test can be done by using body fluids or cells of the patient, this includes; blood (which is the most common), saliva, amniotic fluid and even cells from the interior of the mouth gotten from a buccal swab. This material is then sent to a specialized genetics lab where technicians will examine it, the test results are sent back to the health provider who requested the analysis and results are discussed with the patient.[64]
Direct to consumer testing can be obtained without a medical professional but is not recommended as the consumer loses the opportunity to discuss their decision with an educated professional.[67] According to the National Library of Medicine in the U.S. genetic testing in America costs in the price range of $100-$2000 depending on the type and intricacy of test.[68]
Preventive actions
Genetic testing is important as if a test comes out positive they are more aware of their own personal health and the health of immediate family members.[69] With the help and advice from a medical professional they can take steps to reduce their elevated risk of cancer development through:
- Regular exercise
- A healthy, balanced diet
- Maintaining a healthy weight
- Not smoking
- Staying safe under the sun’s harmful rays [70]
There are other forms of preventive actions, an example for Hereditary Breast and Ovarian Cancer would be to go through surgery: A hysterectomy is the removal of all or some of the uterus, whereas a mastectomy is removing a breast (double mastectomy meaning that both breasts are removed), this can often add years onto their life expectancy.[71] Another preventive measure is regular cancer screening and check-ups. If a person has Lynch’s syndrome then they should have a regular colonoscopy to examine if there is any change in the cells lining the intestinal wall, regular check-ups have been proven to add an average of 7 years onto the life expectancy of a person suffering from Lynch’s syndrome as early detection means the correct preventive actions and surgery can be taken quicker.[72] Regular breast screening is also recommended for women diagnosed with BRCA mutations, as well as that, recent studies show that men with increased risks of developing prostate cancer due to BRCA mutations can decrease their risk by taking aspirin.[73] Aspirin is hugely beneficial in lowering cancer prevalence; however, it must be taken regularly over at least a five-year period to have any effect.[74]
Prevalence of genetic mutations in different ethnic groups
Often genetic mutations are more common in certain ethnic groups, this is because a race can track their ancestors back to one geographic location, the mutated genes are then passed from ancestors down through generations which is why some ethnicities are more susceptible to mutations, thus increasing their chances of developing cancer [61]. As mentioned above, this can be useful as it can help health professionals assess a patient’s risk of having a mutation before they undergo testing.[65] Werner's Syndrome has a prevalence of 1 in 200,000 live births in the U.S., but it affects individuals in Japan in 1 in 20,000-40,000 cases.[75] 1 in 40 Ashkenazi Jews have a BRCA mutation, this is a huge contrast from the general population in the United States where 1 in 400 people are affected. Ashkenazi Jews are at high risk of developing hereditary breast and ovarian cancer and it is recommend that they undergo both genetic testing to see if they have a mutation and regular screening for cancer.[76]
References
- Allgayer, Heike; Redher, Helga; Fulda, Simone (2009). Hereditary Tumors: From Genes to Clinical Consequences. Weinheim: Wiley-VCH. ISBN 9783527320288.
- Hodgson S (January 2008). "Mechanisms of inherited cancer susceptibility". J Zhejiang Univ Sci B. 9 (1): 1–4. doi:10.1631/jzus.B073001. PMC 2170461. PMID 18196605.
- Clark AS, Domchek SM (April 2011). "Clinical management of hereditary breast cancer syndromes". J Mammary Gland Biol Neoplasia. 16 (1): 17–25. doi:10.1007/s10911-011-9200-x. PMID 21360002.
- Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR (July 2009). "Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications". Clin. Genet. 76 (1): 1–18. doi:10.1111/j.1399-0004.2009.01230.x. PMC 2846640. PMID 19659756.
- "Genetics". National Cancer Institute. 2015-04-22. Retrieved 2018-02-20.
- Banks, KC; Moline, JJ; Marvin, ML; Newlin, AC; Vogel, KJ (March 2013). "10 rare tumors that warrant a genetics referral". Familial Cancer. 12 (1): 1–18. doi:10.1007/s10689-012-9584-9. PMID 23377869.
- Korde, Larissa A.; Gadalla, Shahinaz M. (2017-05-02). "Cancer Risk Assessment for the Primary Care Physician". Primary Care. 36 (3): 471–488. doi:10.1016/j.pop.2009.04.006. PMC 2713871. PMID 19616151.
- Anderson, Cindy Lou; Carie A Braun (2007). Pathophysiology: functional alterations in human health. Hagerstwon, MD: Lippincott Williams & Wilkins. ISBN 978-0-7817-6250-2.
- Lindor NM, Greene MH (July 1998). "The concise handbook of family cancer syndromes. Mayo Familial Cancer Program". J. Natl. Cancer Inst. 90 (14): 1039–71. doi:10.1093/jnci/90.14.1039. PMID 9672254.
- Moldovan GL, D'Andrea AD (2009). "How the fanconi anemia pathway guards the genome". Annu. Rev. Genet. 43: 223–49. doi:10.1146/annurev-genet-102108-134222. PMC 2830711. PMID 19686080.
- Tischkowitz MD, Hodgson SV (January 2003). "Fanconi anaemia". Journal of Medical Genetics. 40 (1): 1–10. doi:10.1136/jmg.40.1.1. PMC 1735271. PMID 12525534.
- Kee Y, D'Andrea AD (November 2012). "Molecular pathogenesis and clinical management of Fanconi anemia". J. Clin. Invest. 122 (11): 3799–806. doi:10.1172/JCI58321. PMC 3484428. PMID 23114602.
- Kottemann MC, Smogorzewska A (January 2013). "Fanconi anaemia and the repair of Watson and Crick DNA crosslinks". Nature. 493 (7432): 356–63. Bibcode:2013Natur.493..356K. doi:10.1038/nature11863. PMC 3700363. PMID 23325218.
- Su X, Huang J (September 2011). "The Fanconi anemia pathway and DNA interstrand cross-link repair". Protein Cell. 2 (9): 704–11. doi:10.1007/s13238-011-1098-y. PMC 4875268. PMID 21948210.
- Half E, Bercovich D, Rozen P (2009). "Familial adenomatous polyposis". Orphanet J Rare Dis. 4: 22. doi:10.1186/1750-1172-4-22. PMC 2772987. PMID 19822006.
- Galiatsatos P, Foulkes WD (February 2006). "Familial adenomatous polyposis". Am. J. Gastroenterol. 101 (2): 385–98. PMID 16454848.
- Macrae F, du Sart D, Nasioulas S (2009). "Familial adenomatous polyposis". Best Pract Res Clin Gastroenterol. 23 (2): 197–207. doi:10.1016/j.bpg.2009.02.010. PMID 19414146.
- Petrucelli N, Daly MB, Feldman GL (May 2010). "Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2". Genet. Med. 12 (5): 245–59. doi:10.1097/GIM.0b013e3181d38f2f. PMID 20216074.
- Smith EC (2012). "An overview of hereditary breast and ovarian cancer syndrome". J Midwifery Womens Health. 57 (6): 577–84. doi:10.1111/j.1542-2011.2012.00199.x. PMID 23050669.
- Drescher KM, Sharma P, Lynch HT (2010). "Current hypotheses on how microsatellite instability leads to enhanced survival of Lynch Syndrome patients". Clin. Dev. Immunol. 2010: 1–13. doi:10.1155/2010/170432. PMC 2901607. PMID 20631828.
- Kunkel TA, Erie DA (2005). "DNA mismatch repair". Annu. Rev. Biochem. 74: 681–710. doi:10.1146/annurev.biochem.74.082803.133243. PMID 15952900.
- Kastrinos F, Syngal S (2011). "Inherited colorectal cancer syndromes". Cancer J. 17 (6): 405–15. doi:10.1097/PPO.0b013e318237e408. PMC 3240819. PMID 22157284.
- Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, Hoegerle S, Boedeker CC, Opocher G, Schipper J, Januszewicz A, Eng C (2004). "Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations". JAMA. 292 (8): 943–51. doi:10.1001/jama.292.8.943. PMID 15328326.
- Malkin D (April 2011). "Li-fraumeni syndrome". Genes Cancer. 2 (4): 475–84. doi:10.1177/1947601911413466. PMC 3135649. PMID 21779515.
- Bakry, D (2013). P53 in the Clinic: TP53 Germline Mutations: Genetics of Li–Fraumeni Syndrome. New York: Springer. pp. 167–188. ISBN 978-1-4614-3676-8.
- Birch JM (July 1994). "Familial cancer syndromes and clusters". Br. Med. Bull. 50 (3): 624–39. doi:10.1093/oxfordjournals.bmb.a072913. PMID 7987644.
- Quesnel S, Malkin D (August 1997). "Genetic predisposition to cancer and familial cancer syndromes". Pediatr. Clin. North Am. 44 (4): 791–808. doi:10.1016/s0031-3955(05)70530-7. PMID 9286285.
- Sampson JR, Jones N (2009). "MUTYH-associated polyposis". Best Pract Res Clin Gastroenterol. 23 (2): 209–18. doi:10.1016/j.bpg.2009.03.006. PMID 19414147.
- Manfredi M, Vescovi P, Bonanini M, Porter S (March 2004). "Nevoid basal cell carcinoma syndrome: a review of the literature". Int J Oral Maxillofac Surg. 33 (2): 117–24. doi:10.1054/ijom.2003.0435. PMID 15050066.
- Lo Muzio L (2008). "Nevoid basal cell carcinoma syndrome (Gorlin syndrome)". Orphanet J Rare Dis. 3: 32. doi:10.1186/1750-1172-3-32. PMC 2607262. PMID 19032739.
- Richard, S; Gardie, B; Couvé, S; Gad, S (May 30, 2012). "Von Hippel-Lindau: How a rare disease illuminates cancer biology". Seminars in Cancer Biology. 23 (1): 26–37. doi:10.1016/j.semcancer.2012.05.005. PMID 22659535.
- Henry, Todd; Campell, James; Hawley, Arthur (1969). Todd-Sanford clinical diagnosis by laboratory methods, edited by Israel Davidsohn [and] John Bernard Henry (14th ed.). Philadelphia: Saunders. p. 555. ISBN 978-0-7216-2921-6.
- Wong WT, n E, Agró Coleman HR, et al. (February 2007). "Genotype–phenotype correlation in von Hippel–Lindau disease with retinal angiomatosis". Archives of Ophthalmology. 125 (2): 239–45. doi:10.1001/archopht.125.2.239. PMC 3019103. PMID 17296901. Archived from the original on 2008-12-12. Retrieved 2008-10-22.
- Lehmann AR, McGibbon D, Stefanini M (2011). "Xeroderma pigmentosum". Orphanet J Rare Dis. 6: 70. doi:10.1186/1750-1172-6-70. PMC 3221642. PMID 22044607.
- Niedernhofer LJ, Bohr VA, Sander M, Kraemer KH (2011). "Xeroderma pigmentosum and other diseases of human premature aging and DNA repair: molecules to patients". Mech. Ageing Dev. 132 (6–7): 340–7. doi:10.1016/j.mad.2011.06.004. PMC 3474983. PMID 21708183.
- Keimling M, Volcic M, Csernok A, Wieland B, Dörk T, Wiesmüller L (2011). "Functional characterization connects individual patient mutations in ataxia telangiectasia mutated (ATM) with dysfunction of specific DNA double-strand break-repair signaling pathways". FASEB J. 25 (11): 3849–60. doi:10.1096/fj.11-185546. PMID 21778326.
- Thompson LH, Schild D (2002). "Recombinational DNA repair and human disease". Mutat. Res. 509 (1–2): 49–78. doi:10.1016/s0027-5107(02)00224-5. PMID 12427531.
- Nimonkar AV, Ozsoy AZ, Genschel J, Modrich P, Kowalczykowski SC (2008). "Human exonuclease 1 and BLM helicase interact to resect DNA and initiate DNA repair". Proc. Natl. Acad. Sci. U.S.A. 105 (44): 16906–11. Bibcode:2008PNAS..10516906N. doi:10.1073/pnas.0809380105. PMC 2579351. PMID 18971343.
- German J (1969). "Bloom's syndrome. I. Genetical and clinical observations in the first twenty-seven patients". Am. J. Hum. Genet. 21 (2): 196–227. PMC 1706430. PMID 5770175.
- Nagaraju G, Scully R (2007). "Minding the gap: the underground functions of BRCA1 and BRCA2 at stalled replication forks". DNA Repair (Amst.). 6 (7): 1018–31. doi:10.1016/j.dnarep.2007.02.020. PMC 2989184. PMID 17379580.
- Lancaster JM, Powell CB, Chen LM, Richardson DL (2015). "Society of Gynecologic Oncology statement on risk assessment for inherited gynecologic cancer predispositions". Gynecol. Oncol. 136 (1): 3–7. doi:10.1016/j.ygyno.2014.09.009. PMID 25238946.
- Thompson LH, Hinz JM (2009). "Cellular and molecular consequences of defective Fanconi anemia proteins in replication-coupled DNA repair: mechanistic insights". Mutat. Res. 668 (1–2): 54–72. doi:10.1016/j.mrfmmm.2009.02.003. PMC 2714807. PMID 19622404.
- Alter BP (2003). "Cancer in Fanconi anemia, 1927-2001". Cancer. 97 (2): 425–40. doi:10.1002/cncr.11046. PMID 12518367.
- Meyer LA, Broaddus RR, Lu KH (2009). "Endometrial cancer and Lynch syndrome: clinical and pathologic considerations". Cancer Control. 16 (1): 14–22. doi:10.1177/107327480901600103. PMC 3693757. PMID 19078925.
- Carethers JM, Stoffel EM (2015). "Lynch syndrome and Lynch syndrome mimics: The growing complex landscape of hereditary colon cancer". World J. Gastroenterol. 21 (31): 9253–61. doi:10.3748/wjg.v21.i31.9253. PMC 4541378. PMID 26309352.
- Kastan MB (2008). "DNA damage responses: mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture". Mol. Cancer Res. 6 (4): 517–24. doi:10.1158/1541-7786.MCR-08-0020. PMID 18403632.
- Viktorsson K, De Petris L, Lewensohn R (2005). "The role of p53 in treatment responses of lung cancer". Biochem. Biophys. Res. Commun. 331 (3): 868–80. doi:10.1016/j.bbrc.2005.03.192. PMID 15865943.
- Testa JR, Malkin D, Schiffman JD (2013). "Connecting molecular pathways to hereditary cancer risk syndromes". Am Soc Clin Oncol Educ Book. 33: 81–90. doi:10.1200/EdBook_AM.2013.33.81. PMC 5889618. PMID 23714463.
- Rapp A, Greulich KO (2004). "After double-strand break induction by UV-A, homologous recombination and nonhomologous end joining cooperate at the same DSB if both systems are available". J. Cell Sci. 117 (Pt 21): 4935–45. doi:10.1242/jcs.01355. PMID 15367581.
- Bartkova J, Tommiska J, Oplustilova L, Aaltonen K, Tamminen A, Heikkinen T, Mistrik M, Aittomäki K, Blomqvist C, Heikkilä P, Lukas J, Nevanlinna H, Bartek J (2008). "Aberrations of the MRE11-RAD50-NBS1 DNA damage sensor complex in human breast cancer: MRE11 as a candidate familial cancer-predisposing gene". Mol Oncol. 2 (4): 296–316. doi:10.1016/j.molonc.2008.09.007. PMC 5527773. PMID 19383352.
- Markkanen E, Dorn J, Hübscher U (2013). "MUTYH DNA glycosylase: the rationale for removing undamaged bases from the DNA". Front Genet. 4: 18. doi:10.3389/fgene.2013.00018. PMC 3584444. PMID 23450852.
- Patel SG, Ahnen DJ (2012). "Familial colon cancer syndromes: an update of a rapidly evolving field". Curr Gastroenterol Rep. 14 (5): 428–38. doi:10.1007/s11894-012-0280-6. PMC 3448005. PMID 22864806.
- Chrzanowska KH, Gregorek H, Dembowska-Bagińska B, Kalina MA, Digweed M (2012). "Nijmegen breakage syndrome (NBS)". Orphanet J Rare Dis. 7: 13. doi:10.1186/1750-1172-7-13. PMC 3314554. PMID 22373003.
- Krokan HE, Bjørås M (2013). "Base excision repair". Cold Spring Harb Perspect Biol. 5 (4): a012583. doi:10.1101/cshperspect.a012583. PMC 3683898. PMID 23545420.
- Kuiper RP, Hoogerbrugge N (2015). "NTHL1 defines novel cancer syndrome". Oncotarget. 6 (33): 34069–70. doi:10.18632/oncotarget.5864. PMC 4741436. PMID 26431160.
- Singh DK, Ahn B, Bohr VA (2009). "Roles of RECQ helicases in recombination based DNA repair, genomic stability and aging". Biogerontology. 10 (3): 235–52. doi:10.1007/s10522-008-9205-z. PMC 2713741. PMID 19083132.
- Anbari KK, Ierardi-Curto LA, Silber JS, Asada N, Spinner N, Zackai EH, Belasco J, Morrissette JD, Dormans JP (2000). "Two primary osteosarcomas in a patient with Rothmund-Thomson syndrome". Clin. Orthop. Relat. Res. 378 (378): 213–23. doi:10.1097/00003086-200009000-00032. PMID 10986997.
- Bohr VA (2005). "Deficient DNA repair in the human progeroid disorder, Werner syndrome". Mutat. Res. 577 (1–2): 252–9. doi:10.1016/j.mrfmmm.2005.03.021. PMID 15916783.
- Monnat RJ (2010). "Human RECQ helicases: roles in DNA metabolism, mutagenesis and cancer biology". Semin. Cancer Biol. 20 (5): 329–39. doi:10.1016/j.semcancer.2010.10.002. PMC 3040982. PMID 20934517.
- Menck CF, Munford V (2014). "DNA repair diseases: What do they tell us about cancer and aging?". Genet. Mol. Biol. 37 (1 Suppl): 220–33. doi:10.1590/s1415-47572014000200008. PMC 3983582. PMID 24764756.
- Lehmann AR, McGibbon D, Stefanini M (2011). "Xeroderma pigmentosum". Orphanet J Rare Dis. 6: 70. doi:10.1186/1750-1172-6-70. PMC 3221642. PMID 22044607.
- Oh KS, Imoto K, Emmert S, Tamura D, DiGiovanna JJ, Kraemer KH (2011). "Nucleotide excision repair proteins rapidly accumulate but fail to persist in human XP-E (DDB2 mutant) cells". Photochem. Photobiol. 87 (3): 729–33. doi:10.1111/j.1751-1097.2011.00909.x. PMC 3082610. PMID 21388382.
- Opletalova K, Bourillon A, Yang W, Pouvelle C, Armier J, Despras E, Ludovic M, Mateus C, Robert C, Kannouche P, Soufir N, Sarasin A (2014). "Correlation of phenotype/genotype in a cohort of 23 xeroderma pigmentosum-variant patients reveals 12 new disease-causing POLH mutations". Hum. Mutat. 35 (1): 117–28. doi:10.1002/humu.22462. PMID 24130121.
- "Genetic Testing for Hereditary Cancer Syndromes". National Cancer Institute. 2013-04-22. Retrieved 2018-02-19.
- Foulkes, William D.; Knoppers, Bartha Maria; Turnbull, Clare (January 2016). "Population genetic testing for cancer susceptibility: founder mutations to genomes". Nature Reviews. Clinical Oncology. 13 (1): 41–54. doi:10.1038/nrclinonc.2015.173. ISSN 1759-4782. PMID 26483301.
- Reference, Genetics Home. "What is genetic testing?". Genetics Home Reference. Retrieved 2018-02-20.
- Myers, Melanie F.; Bernhardt, Barbara A. (June 2012). "Direct-to-consumer genetic testing: introduction to the special issue". Journal of Genetic Counseling. 21 (3): 357–360. doi:10.1007/s10897-012-9500-3. ISSN 1573-3599. PMID 22441809.
- Reference, Genetics Home. "What is the cost of genetic testing, and how long does it take to get the results?". Genetics Home Reference. Retrieved 2018-02-20.
- Robson, Mark E.; Bradbury, Angela R.; Arun, Banu; Domchek, Susan M.; Ford, James M.; Hampel, Heather L.; Lipkin, Stephen M.; Syngal, Sapna; Wollins, Dana S. (2015-11-01). "American Society of Clinical Oncology Policy Statement Update: Genetic and Genomic Testing for Cancer Susceptibility". Journal of Clinical Oncology. 33 (31): 3660–3667. doi:10.1200/JCO.2015.63.0996. ISSN 1527-7755. PMID 26324357.
- "Genetic testing for cancer risk". Cancer Research UK. 2015-06-02. Retrieved 2018-02-20.
- Schrag, D.; Kuntz, K. M.; Garber, J. E.; Weeks, J. C. (1997-05-15). "Decision analysis--effects of prophylactic mastectomy and oophorectomy on life expectancy among women with BRCA1 or BRCA2 mutations". The New England Journal of Medicine. 336 (20): 1465–1471. doi:10.1056/NEJM199705153362022. ISSN 0028-4793. PMID 9148160.
- Newton, K.; Green, K.; Lalloo, F.; Evans, D. G.; Hill, J. (January 2015). "Colonoscopy screening compliance and outcomes in patients with Lynch syndrome". Colorectal Disease. 17 (1): 38–46. doi:10.1111/codi.12778. ISSN 1463-1318. PMID 25213040.
- Cossack, Matthew; Ghaffary, Cameron; Watson, Patrice; Snyder, Carrie; Lynch, Henry (April 2014). "Aspirin use is associated with lower prostate cancer risk in male carriers of BRCA mutations". Journal of Genetic Counseling. 23 (2): 187–191. doi:10.1007/s10897-013-9629-8. ISSN 1573-3599. PMID 23881471.
- Thorat, Mangesh A.; Cuzick, Jack (December 2013). "Role of aspirin in cancer prevention". Current Oncology Reports. 15 (6): 533–540. doi:10.1007/s11912-013-0351-3. ISSN 1534-6269. PMID 24114189.
- Reference, Genetics Home. "Werner syndrome". Genetics Home Reference. Retrieved 2018-02-20.
- "Genetic risk, race and ethnicity | Cancer Fighters Thrive Magazine". CancerCenter.com. Archived from the original on 2018-02-21. Retrieved 2018-02-20.