22q11.2 distal deletion syndrome
22q11.2 distal deletion syndrome is a rare genetic condition caused by a tiny missing part of one of the body’s 46 chromosomes – chromosome 22. 22q11.2 distal deletion syndrome appears to be a recurrent genomic disorder distinct from DiGeorge syndrome (DGS; 188400) and velocardiofacial syndrome (VCFS; 192430).[3]
| 22q11.2 distal deletion syndrome | |
|---|---|
| Other names | Distal del(22)(q11.2)[1] |
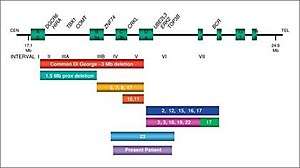 | |
| Schematic overview of the 22q11.2 deletion region [2] | |
The first published description of a person with a 22q11.2 distal deletion was in 1999.[4] There have since been hundreds of cases reported worldwide. 22q11.2 distal deletion syndrome seems to occur equally often in males and females. There are reports of people who are unaffected by carrying the deletion and only discovered it after their child was diagnosed. It seems that the 22q11.2 distal deletion can be "silent" and it is unknown how many people may have a silent form of this syndrome.
Presentation
Every 22q11.2 distal deletion is unique, and each person may have different medical and developmental concerns. A number of common features have emerged:
- Some children are likely to need support with learning. The amount of support needed by each child will vary
- Speech is often delayed and some children have articulation problems
- Growth delay both in the womb and after birth
- Heart problems
- Behavioral difficulties such as difficulties with concentration and anxiety
- Subtly unusual facial features. Families may notice similarities between their own child and others with the deletion
Cause
22q11.2 distal deletion occurs spontaneously; there is no known environmental cause. The genetic term for this is de novo; both parents typically have normal chromosomes. This is hereditary and people affected by distal deletion syndrome have a 50/50 chance of passing it to their children.
De novo 22q11.2 distal deletions are caused by a mistake that is thought to occur when the parents’ sperm or egg cells are formed. At one point in the formation, all the chromosomes including the two chromosome 22s pair up and swap segments. To pair up precisely, each chromosome ‘recognizes’ matching or near-matching DNA sequences on its partner chromosome. However, throughout each chromosome there are many similar DNA sequences that can overlap incorrectly, and pair with the wrong sequence. It is believed that when the exchange of genetic material, known as ‘crossing over’, occurs after mismatching, it is unequal, looping out and excising a length of the chromosome.
Diagnosis
Treatment
Research
The features of 22q11.2 distal deletion syndrome are likely to be the result of the loss of a number of different genes found in this region. Most people have an approximately 0.4 to 2.1 Mb deletion (400'000- 2. Millions bases). Although the gene(s) responsible for the clinical features associated with 22q11.2 distal deletion syndrome have not been clearly defined, several potential candidate genes have been suggested.
CRKL and MAPK1 genes have been suggested to have a role in the heart anomalies that are common in 22q11.2 distal deletion syndrome. MAPK1 has also been suggested to be associated with placental development and therefore having one copy of this gene missing in 22q11.2 distal deletion syndrome may be linked to the tendency for premature birth and IUGR.[5] The MAPK1 gene in mice has been shown to contribute to social behavior[6] and therefore may play a role in the behavioral problems found in some people with 22q11.2 distal deletion syndrome.
Very distal deletions including the SMARCB1 gene are associated with an increased risk of malignant rhabdoid tumors. Very little is known about the magnitude of the risk for malignancy associated with distal 22q11.2 deletion syndrome but it is advised that people with a deletion that includes the SMARCB1 gene undergo careful, prolonged monitoring for this type of tumor. Most persons with 22q11 distal deletions do not have deletion of the SMARCB1 gene.
See also
- 22q11.2 deletion syndrome
References
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Distal 22q11.2 microdeletion syndrome". www.orpha.net. Retrieved 26 October 2019.
- Garavelli, L.; Rosato, S.; Wischmeijer, A.; Gelmini, C.; Esposito, A.; Mazzanti, L.; Franchi, F.; De Crescenzo, A.; Palumbo, O.; Carella, M.; Riccio, A. (2011). "22q11.2 Distal Deletion Syndrome: Description of a New Case with Truncus Arteriosus Type 2 and Review". Mol Syndromol. 2 (1): 35–44. doi:10.1159/000334262. PMC 3343753. PMID 22582037.
- Ben-Shachar, S; Ou Z; Shaw CA; Belmont JW; Patel MS; Hummel M; Amato S; Tartaglia N; Berg J; Sutton VR; Lalani SR; Chinault AC; Cheung SW; Lupski JR; Patel A (Jan 2008). "22q11.2 distal deletion: a recurrent genomic disorder distinct from DiGeorge syndrome and velocardiofacial syndrome". Am J Hum Genet. 82 (1): 214–21. doi:10.1016/j.ajhg.2007.09.014. PMC 2253964. PMID 18179902.
- Saitta, SC; McGrath JM; Mensch H; Shaikh TH; Zackai EH; Emanuel BS Am J Hum Genet (August 1999). "A 22q11.2 deletion that excludes UFD1L and CDC45L in a patient with conotruncal and craniofacial defects". Am J Hum Genet. 65 (2): 562–6. doi:10.1086/302514. PMC 1377955. PMID 10417299.
- Fagberg, CR; Graakjaer J; Heinl UD; Ousager LB; Dreyer I; Kirchhoff M; Rasmussen AA; Lautrup CK; Birkebaek N; Sorensen K. (February 2013). "Heart defects and other features of the 22q11 distal deletion syndrome". Eur J Med Genet. 56 (2): 98–107. doi:10.1016/j.ejmg.2012.09.009. PMID 23063575.
- Satoh, Y; Endo S; Nakata T; Kobayashi Y; Yamada K; Ikeda T; Takeuchi A; Hiramoto T; Watanabe Y; Kazama T (Aug 2011). "ERK2 contributes to the control of social behaviors in mice". J Neurosci. 31 (33): 11953–67. doi:10.1523/JNEUROSCI.2349-11.2011. PMC 6623182. PMID 21849556.