Williams syndrome
Williams syndrome (WS) is a genetic disorder that affects many parts of the body.[3] Facial features frequently include a broad forehead, short nose and full cheeks, an appearance that has been described as "elfin".[3][5] While mild to moderate intellectual disability with particular problems with visual spatial tasks such as drawing is typical, verbal skills are generally relatively unaffected.[3] Those affected often have an outgoing personality, interact readily with strangers, and appear happy.[3][5] Problems with teeth, heart problems (especially supravalvular aortic stenosis), and periods of high blood calcium are common.[2][3]
| Williams syndrome | |
|---|---|
| Other names | Williams–Beuren syndrome (WBS) |
.jpg) | |
| Two men, age 21 and 28, displaying several of the facial features characteristic of Williams syndrome.[1] | |
| Specialty | Medical genetics, pediatrics |
| Symptoms | Facial changes, intellectual disability, overly friendly nature, short height[2] |
| Complications | Heart problems, periods of high blood calcium[2][3] |
| Duration | Lifelong[2] |
| Causes | Genetic[2] |
| Differential diagnosis | Noonan syndrome, fetal alcohol syndrome, DiGeorge syndrome[2] |
| Treatment | Various types of therapy[2] |
| Prognosis | Shorter life expectancy[4] |
| Frequency | 1 in 7,500 to 1 in 20,000[5] |
Williams syndrome is caused by a genetic abnormality, specifically a deletion of about 27 genes from the long arm of one of the two chromosome 7s.[3][5] Typically this occurs as a random event during the formation of the egg or sperm from which a person develops.[3] In a small number of cases, it is inherited from an affected parent in an autosomal dominant manner.[3] The different characteristic features have been linked to the loss of specific genes.[3] The diagnosis is typically suspected based on symptoms and confirmed by genetic testing.[2]
Treatment includes special education programs and various types of therapy.[2] Surgery may be done to correct heart problems.[2] Dietary changes or medications may be required for high blood calcium.[2] The syndrome was first described in 1961 by New Zealander John C. P. Williams.[6][7] Williams syndrome affects between 1 in 7,500 to 1 in 20,000 people at birth.[5] Life expectancy is less than that of the general population, mostly due to the increased rates of heart disease.[4]
Signs and symptoms
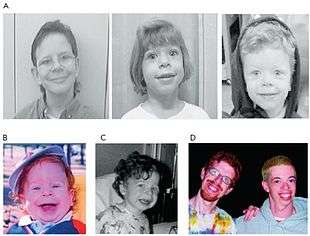
The most common symptoms of Williams syndrome are heart defects and unusual facial features. Other symptoms include failure to gain weight appropriately in infancy (failure to thrive) and low muscle tone. People with Williams syndrome tend to have widely spaced teeth, a long philtrum, and a flattened nasal bridge.[8]
Most people with Williams syndrome are highly verbal relative to their IQ, and are often very sociable, having what has been described as a "cocktail party" type personality.[9] People with Williams syndrome hyperfocus on the eyes of others in social engagements.[10]
Physical
People with Williams syndrome experience many cardiac problems, commonly heart murmurs and the narrowing of major blood vessels as well as supravalvular aortic stenosis. Other symptoms may include gastrointestinal problems, such as severe or prolonged colic,[11] abdominal pain and diverticulitis, nocturnal enuresis (bed wetting) and urinary difficulties, dental irregularities and defective tooth enamel, as well as hormone problems, the most common being high blood calcium.[12] Hypothyroidism has been reported to occur in children, although there is no proof of it occurring in adults; adults with WS have a higher risk of developing type 2 diabetes, with some cases apparent as young as 21 years old.[13]
People with Williams syndrome often have hyperacusia and phonophobia which resembles noise-induced hearing loss, but this may be due to a malfunctioning auditory nerve.[14][15] However, people with WS can also tend to demonstrate a love of music,[7] and they appear significantly more likely to possess absolute pitch.[16] There also appears to be a higher prevalence of left-handedness and left-eye dominance.[17]
Ophthalmologic issues are common in Williams syndrome. Up to 75% of subjects in some studies have strabismus (ocular misalignment), particularly esotropia,[18] due to inherent subnormal binocular visual function[19] and cognitive deficits in visuospatial construction.[20] People with Williams syndrome have problems with visual processing, but this is related to difficulty in dealing with complex spatial relationships rather than depth perception per se.[21]
Nervous system
Because of the multiple genes that are missing in people with Williams syndrome, there are many effects on the brain, including abnormalities in the cerebellum, right parietal lobe, and left frontal cortical regions. This pattern is consistent with the visual-spatial disabilities and problems with behavioral timing often seen in Williams syndrome.
Frontal-cerebellar pathways, involved in behavioral timing, are often abnormally developed in people with Williams syndrome, which may be related to their deficits in coordination and execution of fine motor tasks such as drawing and writing. In addition, people with Williams syndrome often exhibit gross motor difficulties, including trouble walking downstairs, as well as overactive motor reflexes (hyperreflexia) and hyperactive, involuntary movement of the eyes (nystagmus).[22]
Williams syndrome is also noteworthy for exhibiting abnormalities in the parietal-dorsal areas of the neocortex, but not the ventral areas. The parietal-dorsal area handles visual processing that supports visual-spatial analysis of the environment, while the ventral is related to semantic recognition of visual stimuli, as well as the recognition of faces. Thus, people with Williams syndrome are often able to visually identify and recognize whole objects, and refer to them by name, but struggle with visuospatial construction (seeing an object as being composed of many smaller parts, and recreating it) and orienting themselves in space.[22]
People with Williams syndrome are often affable and hyperverbal, demonstrating the decreased inhibition ability that stems from dorsal-frontal deficits.[23] Some studies suggest that the amygdala of a person with Williams syndrome has greater volume than the average person's (though it is smaller than average in childhood).[24] In general, neuroimaging studies demonstrate that people with Williams syndrome have diminished amygdala reactivity in response to socially frightening stimuli (such as disapproving faces), but demonstrate hyperreactivity in the amygdala when presented with nonsocial fear stimuli (such as frightening animals).[22] This may partially account for the apparent absence of social inhibition observed in people with the syndrome, as well as the prevalence of anxious symptoms (but see fear for details on the relationship between the amygdala and fear response).[25] There is also evidence that people with Williams exhibit hyper amygdala activity when viewing happy facial expressions.[26]
Increased volume and activation of the left auditory cortex has been observed in people with Williams syndrome, which has been interpreted as a neural correlate of patients' rhythm propensity and fondness of music. Similar sizes of the auditory cortex have been previously reported only in professional musicians.[27]
Developmental
The earliest observable symptoms of Williams syndrome include low birth weight, failure to thrive, trouble breastfeeding, nocturnal irritability and gastroesophageal reflux. Facial dysmorphies thought to be characteristic of the syndrome are also present early in development, as is a heart murmur. Research on the development of the syndrome suggests that congenital heart disease is typically present at an early age, often at the infant's first pediatric appointment. Heart problems in infancy often lead to the initial diagnosis of Williams syndrome.[24]
Developmental delays are present in most cases of Williams syndrome, and include delay of language abilities and delayed motor skill development. People with Williams syndrome develop language abilities quite late relative to other children, with the child's first word often occurring as late as three years of age. Language abilities are often observed to be deficient until adolescence, in terms of semantics, morphology, and phonology, though not in vocabulary.[24]
Williams syndrome is also marked by a delay in the development of motor skills. Infants with Williams develop the ability to lift their heads and sit without support months later than typically developing children. These delays continue into childhood, where patients with Williams syndrome are delayed in learning to walk.[24] In young children, the observed motor delay is around five to six months, though some research suggests that children with Williams syndrome have a delay in development that becomes more extreme with age.[28] Children with motor delays as a result of Williams syndrome are particularly behind in the development of coordination, fine motor skills such as writing and drawing, response time, and strength and dexterity of the arms. Impaired motor ability persists (and possibly worsens) as children with Williams syndrome reach adolescence.[29]
Adults and adolescents with Williams syndrome typically achieve a below-average height and weight, compared with non-affected populations. As people with Williams syndrome age, they frequently develop joint limitations and hypertonia, or abnormally increased muscle tone. Hypertension, gastrointestinal problems, and genitourinary symptoms often persist into adulthood, as well as cardiovascular problems. Adults with Williams syndrome are typically limited in their ability to live independently or work in competitive employment settings, but this developmental impairment is attributed more to psychological symptoms than physiological problems.[30]
Social and psychological
People with Williams syndrome report higher anxiety levels as well as phobia development, which may be associated with hyperacusis (high sensitivity to certain frequencies of sound).[31] Compared with other children with delays, those with Williams syndrome display a significantly greater number of fears. 35% of these children met the DSM definition of having a phobia as compared with 1–4.3% for those with other types of developmental delays.[32] Williams syndrome is also strongly associated with attention deficit hyperactivity disorder and related psychological symptoms such as poor concentration, hyperactivity, and social disinhibition.[9]
Furthermore, cognitive abilities (IQs) of people with WMS typically range from mild-to-moderate levels of intellectual disability.[33] One study of 306 children with Williams syndrome found IQ scores ranging from 40 to 112 with a mean of 69.32 (an IQ score of 100 is by definition the average in any single large enough and generally homogeneous population).[34] IQ scores above this range have been reported in people with smaller genetic deletions.[13] In particular, people with Williams syndrome experience challenges in visual-motor skills and visuospatial construction. Most affected people are unable to spatially orient themselves and many experience difficulty when given a task that requires even the most basic visual problem-solving. Many adults with Williams syndrome cannot complete a simple six-piece puzzle designed for young children, for example. These visuospatial deficits may be related to damage to the dorsal cortical pathway for visual processing.[35]
Despite their physical and cognitive deficits, people with Williams syndrome exhibit impressive social and verbal abilities. Williams patients can be highly verbal relative to their IQ. When children with Williams syndrome are asked to name an array of animals, they may well list a wild assortment of creatures such as a koala, saber-toothed cat, vulture, unicorn, sea lion, yak, ibex and Brontosaurus, a far greater verbal array than would be expected of children with IQs in the 60s.[36] Some other strengths that have been associated with Williams syndrome are auditory short-term memory and facial recognition skills. The language used by people with Williams syndrome differs notably from unaffected populations, including people matched for IQ. People with Williams syndrome tend to use speech that is rich in emotional descriptors, high in prosody (exaggerated rhythm and emotional intensity), and features unusual terms and strange idioms.[35]
Among the hallmark traits of people with Williams syndrome is an apparent lack of social inhibition. Dykens and Rosner (1999) found that 100% of those with Williams syndrome were kind-spirited, 90% sought the company of others, 87% empathize with others' pain, 84% are caring, 83% are unselfish/forgiving, 75% never go unnoticed in a group, and 75% are happy when others do well.[37] Infants with Williams syndrome make normal and frequent eye contact, and young children with Williams will often approach and hug strangers. People affected by Williams syndrome typically have high empathy and are rarely observed displaying aggression. In regards to empathy, they show relative strength in reading people's eyes to gauge intentions, emotions, and mental states.[38] The level of friendliness observed in people with Williams is often inappropriate for the social setting, however, and teens and adults with Williams syndrome often experience social isolation, frustration, and loneliness despite their clear desire to connect to other people.[35]
While these children often came off as happy due to their sociable nature, often there are internal drawbacks to the way they act. 76–86% of these children were reported as believing that they either had few friends or problems with their friends. This is possibly due to the fact that although they are very friendly to strangers and love meeting new people, they may have trouble interacting on a deeper level. 73–93% were reported as unreserved with strangers, 67% highly sensitive to rejection, 65% susceptible to teasing, and the statistic for exploitation and abuse was unavailable.[37][39][40][41][42] This last one is a significant problem. People with Williams syndrome are frequently very trusting and want more than anything to make friends, leading them to submit to requests that under normal circumstances would be rejected. There are external problems as well. 91–96% demonstrate inattention, 75% impulsivity, 59–71% hyperactivity 46–74% tantrums, 32–60% disobedience, and 25–37% fighting and aggressive behavior.[37][39][43]
In one experiment, a group of children with Williams syndrome showed no signs of racial bias, unlike children without the syndrome. They did show gender bias, however, to a similar degree to children without the syndrome, suggesting separate mechanisms for these biases.[44]
Cause
Williams syndrome is a microdeletion syndrome caused by the spontaneous deletion of genetic material from the region q11.23 of one member of the pair of chromosome 7, so that the person is hemizygous for those genes.[47][48] The deleted region includes more than 25 genes, and researchers believe that being hemizygous for these genes probably contributes to the characteristic features of this syndrome. CLIP2, ELN, GTF2I, GTF2IRD1, and LIMK1 are among the genes that are typically deleted from one chromosome in people with Williams syndrome. Researchers have found this hemizygosity for the ELN gene, which codes for the protein elastin, is associated with the connective-tissue abnormalities and cardiovascular disease (specifically supravalvular aortic stenosis and supravalvular pulmonary stenosis) found in many people with this syndrome. The insufficient supply of elastin may also be the cause of full cheeks, harsh or hoarse voice, hernias and bladder diverticula often found in those with Williams syndrome. Studies suggest that hemizygosity in LIMK1, GTF2I, GTF2IRD1, and perhaps other genes may help explain the characteristic difficulties with visual–spatial tasks. Additionally, there is evidence that the hemizygosity in several of these genes, including CLIP2, may contribute to the unique behavioral characteristics, learning disabilities, and other cognitive difficulties seen in Williams syndrome.[49]
Diagnosis
According to the Williams Syndrome Association, diagnosis of Williams syndrome begins with the recognition of physical symptoms and markers, which is followed by a confirmatory genetic test. The physical signs that often indicate a suspected case of Williams syndrome include puffiness around the eyes, a long philtrum, and a stellate pattern in the iris. Physiological symptoms that often contribute to a Williams syndrome diagnosis are cardiovascular problems, particularly aortic or pulmonary stenosis, as well as feeding disturbance in infants. Developmental delays are often taken as an initial sign of the syndrome, as well.[50]
If a physician suspects a case of Williams syndrome, the diagnosis is confirmed using one of two possible genetic tests: micro-array analysis or the fluorescent in situ hybridization (FISH) test. The FISH test examines chromosome #7 and probes for the existence of two copies of the elastin gene. Since 98-99% of individuals with Williams syndrome lack half of the 7q11.23 region of chromosome #7, where the elastin gene is located, the presence of only one copy of the gene is a strong sign of the syndrome.[50] This confirmatory genetic test has been validated in epidemiological studies of the syndrome and has been demonstrated to be a more effective method of identifying Williams syndrome than previous methods, which often relied on the presence of cardiovascular problems and facial features (which, while common, are not always present).[51]
Some diagnostic studies suggest that reliance on facial features to identify Williams syndrome may cause a misdiagnosis of the condition. Among the more reliable features suggestive of Williams are congenital heart disease, periorbital fullness ("puffy" eyes), and the presence of a long smooth philtrum. Less reliable signs of the syndrome include anteverted nostrils, a wide mouth, and an elongated neck. Researchers indicate that even with significant clinical experience, it is difficult to reliably identify Williams syndrome based on facial features alone.[24]
Treatment
There is no cure for Williams syndrome. Suggestions include avoidance of extra calcium and vitamin D, as well as treating high levels of blood calcium. Blood vessel narrowing can be a significant health problem and is treated on an individual basis. Physical therapy is helpful to patients with joint stiffness and low muscle tone. Developmental and speech therapy can also help children and increase the success of their social interactions. Other treatments are based on a patient's particular symptoms.[8]
The American Academy of Pediatrics recommends annual cardiology evaluations for individuals with Williams syndrome. Other recommended assessments include: ophthalmologic evaluations, an examination for inguinal hernia, objective hearing assessment, blood pressure measurement, developmental and growth evaluation, orthopedic assessments on joints, muscle tone, and ongoing feeding and dietary assessments to manage constipation and urinary problems.[52]
Behavioral treatments have been shown to be effective. In regards to social skills, it may be effective to channel their nature by teaching basic skills. Some of these are the appropriate way to approach someone, how and when to socialize in settings such as school or the workplace, and warning of the signs and dangers of exploitation. For the fear that they demonstrate cognitive-behavioral approaches, such as therapy, are the recommended treatment. One of the things to be careful of with this approach is to make sure that the patients' charming nature does not mask any underlying feelings.
Perhaps the most effective treatment for those with Williams syndrome is music. Those with Williams syndrome have shown relative strength in regards to music, albeit only in pitch and rhythm tasks. Not only do they show strength in the field but also a particular fondness for it. It has been shown that music may help with the internal and external anxiety that these people are more likely to be afflicted with.[53] Something of note is that the typical person processes music in the superior temporal and middle temporal gyri. Those with Williams syndrome have reduced activation in these areas but an increase in the right amygdala and cerebellum.
People affected by Williams syndrome are supported by multiple organizations, including the Canadian Association for Williams Syndrome and the Williams Syndrome Registry.[54]
Epidemiology
Williams syndrome has historically been estimated to occur in roughly 1 in every 20,000 live births.[22] However, more recent epidemiological studies have placed the occurrence rate at closer to 1 in every 7,500 live births, a significantly larger prevalence. As an increasing body of evidence suggests that Williams syndrome is more common than originally noted (approximately 6% of all genetic cases of developmental disability), researchers have begun to theorize past under-diagnosis of the syndrome.[55] One theorized reason for the increase in epidemiological estimates is that there exists a substantial minority of individuals with the genetic markers of Williams syndrome who lack the characteristic facial features or the diminished IQ considered to be diagnostic of the syndrome, who often are not immediately recognized as people with the syndrome.[9][56]
History
Williams syndrome was first described by J. C. P. Williams and his colleagues, who wrote in 1961 of four patients with supravalvular aortic stenosis, mental disability, and facial features including a broad forehead, large chin, low-set, "drooping" cheeks, widely spaced eyes, and a wide-set mouth. A year after this report, German physician A. J. Beuren described three new patients with the same presentation. This led to the syndrome's full original name of Williams-Beuren syndrome, which is still used in some medical publications. From 1964 to 1975, small research reports broadened medical knowledge of this syndrome's cardiovascular problems. Then in 1975, K. Jones and D. Smith conducted a large-scale report on numerous patients with Williams syndrome, ranging in age from infancy to adulthood, and described the behavioral and observable physical symptoms in greater detail than previously recorded.[9]
Society and culture

The adjective "elfin" may have originated to describe the facial features of people with Williams syndrome; before Williams syndrome's scientific cause was understood, people believed that individuals with the syndrome, who have exceptionally charming and kind personalities, had extraordinary, even magical, powers. This has been proposed to be the origin of the folklore of elves, fairies and other forms of the 'good people' or 'wee folk' present in English folklore.[57]
In a review of the symptoms and features of the syndrome, Laskari, Smith and Graham emphasized that some family members of individuals with Williams syndrome typically reject use of terminology such as "elfin", as well as descriptions of social symptoms as "cocktail party syndrome". Physicians, family members of individuals with Williams syndrome, and Williams syndrome associations alike have called for the curtailment of such terms.[9]
One notable person with the syndrome is Gabrielle Marion-Rivard, a Canadian actress and singer who won the Canadian Screen Award for Best Actress in 2014 for her performance in the film Gabrielle.[58] Another is Jeremy Vest,[59] member of the How's Your News? team, featured in the TV series and film of the same name.
References
- Nikitina, EA; Medvedeva, AV; Zakharov, GA; Savvateeva-Popova, EV (January 2014). "Williams syndrome as a model for elucidation of the pathway genes - the brain - cognitive functions: genetics and epigenetics". Acta Naturae. 6 (1): 9–22. doi:10.32607/20758251-2014-6-1-9-22. PMC 3999462. PMID 24772323.
- Morris, CA; Pagon, RA; Adam, MP; Ardinger, HH; Wallace, SE; Amemiya, A; Bean, LJH; Bird, TD; Ledbetter, N; Mefford, HC; Smith, RJH; Stephens, K (2013). "Williams Syndrome". GeneReviews. PMID 20301427.
- Reference, Genetics Home (December 2014). "Williams syndrome". Genetics Home Reference. Archived from the original on 20 January 2017. Retrieved 22 January 2017.

- Riccio, Cynthia A.; Sullivan, Jeremy R.; Cohen, Morris J. (2010). Neuropsychological Assessment and Intervention for Childhood and Adolescent Disorders. John Wiley & Sons. p. 400. ISBN 9780470570333.
- Martens, Marilee A.; Wilson, Sarah J.; Reutens, David C. (2008). "Research Review: Williams syndrome: A critical review of the cognitive, behavioral, and neuroanatomical phenotype". Journal of Child Psychology and Psychiatry. 49 (6): 576–608. doi:10.1111/j.1469-7610.2008.01887.x. PMID 18489677.
- Lenhoff, Howard M.; Teele, Rita L.; Clarkson, Patricia M.; Berdon, Walter E. (2010). "John C. P. Williams of Williams-Beuren syndrome". Pediatric Radiology. 41 (2): 267–9. doi:10.1007/s00247-010-1909-y. PMID 21107555.
- Dobbs, David (2007-07-08). "The Gregarious Brain". New York Times. Archived from the original on 2008-12-11. Retrieved 2007-09-25.
- Jasmin, L. (2009-10-14). "Williams syndrome". MedlinePlus. Archived from the original on 2011-12-01. Retrieved 2011-12-07.
- Lashkari, A.; Smith, A. K.; Graham, J. M. (1999). "Williams-Beuren Syndrome: An Update and Review for the Primary Physician". Clinical Pediatrics. 38 (4): 189–208. doi:10.1177/000992289903800401. PMID 10326175.
- Riby, Deborah M.; Hancock, Peter J.B. (2008). "Viewing it differently: Social scene perception in Williams syndrome and Autism" (PDF). Neuropsychologia. 46 (11): 2855–60. doi:10.1016/j.neuropsychologia.2008.05.003. hdl:1893/468. PMID 18561959.
- "Williams Syndrome". CHD-UK. Archived from the original on 2011-12-11. Retrieved 2011-12-07.
- Cassidy SB; Allanson JE (5 April 2010). Management of Genetic Syndromes. John Wiley and Sons. pp. 909–23. ISBN 978-0-470-19141-5. Retrieved 7 December 2011.
- Morris, Colleen; Lenhoff, Howard; Wang, Paul (2006). Williams-Beuren Syndrome: Research, Evaluation, and Treatment. Johns Hopkins University Press. pp. 70–71. ISBN 978-0-8018-8212-8.
- Gothelf, D.; Farber, N.; Raveh, E.; Apter, A.; Attias, J. (2006). "Hyperacusis in Williams syndrome: Characteristics and associated neuroaudiologic abnormalities". Neurology. 66 (3): 390–5. CiteSeerX 10.1.1.545.5502. doi:10.1212/01.wnl.0000196643.35395.5f. PMID 16476938.
- Johnson, Liane B.; Comeau, Michel; Clarke, Kevin D. (2001). "Hyperacusis in Williams Syndrome". The Journal of Otolaryngology. 30 (2): 90–2. doi:10.2310/7070.2001.20811. PMID 11770962.
- Sacks, O.; Schlaug, G.; Jancke, L.; Huang, Y.; Steinmetz, H. (1995). "Musical ability". Science. 268 (5211): 621–2. Bibcode:1995Sci...268..621S. doi:10.1126/science.7732360. PMID 7732360.
- Van Strien, J. W.; Lagers-Van Haselen, G. C.; Van Hagen, J. M.; De Coo, I. F. M.; Frens, M. A.; Van Der Geest, J. N. (2007). "Increased Prevalences of Left-handedness and Left-eye Sighting Dominance in People with Williams-Beuren Syndrome". Journal of Clinical and Experimental Neuropsychology. 27 (8): 967–76. doi:10.1080/13803390490919119. PMID 16207621.
- Kapp, M. E.; von Noorden, G. K.; Jenkins, R (1995). "Strabismus in Williams syndrome". American Journal of Ophthalmology. 119 (3): 355–60. doi:10.1016/s0002-9394(14)71180-8. PMID 7503839.
- Olitsky, S. E.; Sadler, L. S.; Reynolds, J. D. (1997). "Subnormal binocular vision in the Williams syndrome". Journal of Pediatric Ophthalmology and Strabismus. 34 (1): 58–60. PMID 9027682.
- Olsen, R. K.; Kippenhan, J. S.; Japee, S.; Kohn, P.; Mervis, C. B.; Saad, Z. S.; Morris, C. A.; Meyer-Lindenberg, A.; Berman, K. F. (2009). "Retinotopically defined primary visual cortex in Williams syndrome". Brain. 132 (3): 635–44. doi:10.1093/brain/awn362. PMC 2724925. PMID 19255058.
- Van Der Geest, J. N.; Lagers-Van Haselen, G. C.; Van Hagen, J. M.; Brenner, E.; Govaerts, L. C. P.; De Coo, I. F. M.; Frens, M. A. (2005). "Visual depth processing in Williams–Beuren syndrome". Experimental Brain Research. 166 (2): 200–9. doi:10.1007/s00221-005-2355-1. PMID 15965761.
- Meyer-Lindenberg, Andreas; Mervis, Carolyn B.; Faith Berman, Karen (2006). "Neural mechanisms in Williams syndrome: A unique window to genetic influences on cognition and behaviour". Nature Reviews Neuroscience. 7 (5): 380–93. doi:10.1038/nrn1906. PMID 16760918.
- Cozolino LJ (17 October 2006). The neuroscience of human relationships: attachment and the developing social brain. Norton. pp. 289–91. ISBN 978-0-393-70454-9. Retrieved 7 December 2011.
- Carrasco, Ximena; Castillo, Silvia; Aravena, Teresa; Rothhammer, Paula; Aboitiz, Francisco (2005). "Williams syndrome: Pediatric, neurologic, and cognitive development". Pediatric Neurology. 32 (3): 166–72. doi:10.1016/j.pediatrneurol.2004.09.013. PMID 15730896.
- Järvinen-Pasley, Anna; Bellugi, Ursula; Reilly, Judy; Mills, Debra L.; Galaburda, Albert; Reiss, Allan L.; Korenberg, Julie R. (2008). "Defining the social phenotype in Williams syndrome: A model for linking gene, the brain, and behavior". Development and Psychopathology. 20 (1): 1–35. doi:10.1017/S0954579408000011. PMC 2892602. PMID 18211726.
- Haas, B. W.; Mills, D; Yam, A; Hoeft, F; Bellugi, U; Reiss, A (2009). "Genetic influences on sociability: Heightened amygdala reactivity and event-related responses to positive social stimuli in Williams syndrome". Journal of Neuroscience. 29 (4): 1132–9. doi:10.1523/JNEUROSCI.5324-08.2009. PMC 2754840. PMID 19176822.
- Wengenroth, Martina; Blatow, Maria; Bendszus, Martin; Schneider, Peter (2010). "Leftward Lateralization of Auditory Cortex Underlies Holistic Sound Perception in Williams Syndrome". PLoS ONE. 5 (8): e12326. Bibcode:2010PLoSO...512326W. doi:10.1371/journal.pone.0012326. PMC 2925895. PMID 20808792.
- Bruno, E; Rossi, N; Thüer, O; Córdoba, R; Alday, L. E. (2003). "Cardiovascular findings, and clinical course, in patients with Williams syndrome". Cardiology in the Young. 13 (6): 532–6. doi:10.1017/S1047951103001124. PMID 14982294.
- Tsai, Sen-Wei; Wu, Shyi-Kuen; Liou, Ying-Ming; Shu, San-Ging (2008). "Early development in Williams syndrome". Pediatrics International. 50 (2): 221–4. doi:10.1111/j.1442-200X.2008.02563.x. PMID 18353064.
- Morris, Colleen A.; Demsey, Susan A.; Leonard, Claire O.; Dilts, Constance; Blackburn, Brent L. (1988). "Natural history of Williams syndrome: Physical characteristics". The Journal of Pediatrics. 113 (2): 318–26. doi:10.1016/s0022-3476(88)80272-5. PMID 2456379.
- Blomberg, S; Rosander, M; Andersson, G (2006). "Fears, hyperacusis and musicality in Williams syndrome". Research in Developmental Disabilities. 27 (6): 668–80. doi:10.1016/j.ridd.2005.09.002. PMID 16269236.
- Dykens, Elisabeth M. (2003). "Anxiety, Fears, and Phobias in Persons with Williams Syndrome". Developmental Neuropsychology. 23 (1–2): 291–316. doi:10.1080/87565641.2003.9651896. PMID 12730029.
- Bellugi, U., Lichtenberger, L., Jones, W., Lai, Z., & George, M. S. (2000). I. The neurocognitive profile of Williams Syndrome: a complex pattern of strengths and weaknesses. Journal of cognitive neuroscience, 12(Supplement 1), 7-29. Chicago
- Mervis, Carolyn B.; Becerra, Angela M. (2007). "Language and communicative development in Williams syndrome". Mental Retardation and Developmental Disabilities Research Reviews. 13 (1): 3–15. doi:10.1002/mrdd.20140. PMID 17326109.
- Kaplan, P.; Wang, P. P.; Francke, U. (2001). "Williams (Williams Beuren) Syndrome: A Distinct Neurobehavioral Disorder". Journal of Child Neurology. 16 (3): 177–90. doi:10.1177/088307380101600305. PMID 11305686.
- Pinker S (4 September 2007). The Language Instinct: How the Mind Creates Language. HarperCollins. p. 53. ISBN 978-0-06-133646-1. Archived from the original on 1 January 2014. Retrieved 7 December 2011.
- Dykens, Elisabeth M.; Rosner, Beth A. (1999). "Refining Behavioral Phenotypes: Personality—Motivation in Williams and Prader-Willi Syndromes". American Journal on Mental Retardation. 104 (2): 158–69. doi:10.1352/0895-8017(1999)104<0158:RBPPIW>2.0.CO;2. PMID 10207579.
- Tager-Flusberg, H; Sullivan, K (2000). "A componential view of theory of mind: Evidence from Williams syndrome". Cognition. 76 (1): 59–90. doi:10.1016/s0010-0277(00)00069-x. PMID 10822043.
- Udwin, Orlee; Yule, William; Martin, Neil (1987). "Cognitive Abilities and Behavioural Characteristics of Children with Idiopathic Infantile Hypercalcaemia". Journal of Child Psychology and Psychiatry. 28 (2): 297–309. doi:10.1111/j.1469-7610.1987.tb00212.x. PMID 3584299.
- Udwin, O (1990). "A survey of adults with Williams syndrome and idiopathic infantile hypercalcaemia". Developmental Medicine & Child Neurology. 32 (2): 129–41. doi:10.1111/j.1469-8749.1990.tb16912.x. PMID 2338177.
- Gosch, A; Pankau, R (1997). "Personality characteristics and behaviour problems in individuals of different ages with Williams syndrome". Developmental Medicine & Child Neurology. 39 (8): 527–33. doi:10.1111/j.1469-8749.1997.tb07481.x. PMID 9295848.
- Howlin, P; Davies, M; Udwin, O (1998). "Cognitive functioning in adults with Williams syndrome". Journal of Child Psychology and Psychiatry, and Allied Disciplines. 39 (2): 183–9. doi:10.1111/1469-7610.00312. PMID 9669231.
- Einfeld, Stewart L.; Tonge, Bruce J.; Florio, Tony (1997). "Behavioral and Emotional Disturbance in Individuals with Williams Syndrome". American Journal on Mental Retardation. 102 (1): 45–53. doi:10.1352/0895-8017(1997)102<0045:BAEDII>2.0.CO;2. PMID 9241407.
- Santos, Andreia; Meyer-Lindenberg, Andreas; Deruelle, Christine (2010). "Absence of racial, but not gender, stereotyping in Williams syndrome children". Current Biology. 20 (7): R307–8. doi:10.1016/j.cub.2010.02.009. PMID 20392417.
- Merla, Giuseppe; Howald, Cédric; Henrichsen, Charlotte N.; Lyle, Robert; Wyss, Carine; Zabot, Marie-Thérèse; Antonarakis, Stylianos E.; Reymond, Alexandre (2006). "Submicroscopic Deletion in Patients with Williams-Beuren Syndrome Influences Expression Levels of the Nonhemizygous Flanking Genes". The American Journal of Human Genetics. 79 (2): 332–41. doi:10.1086/506371. PMC 1559497. PMID 16826523.
- Schubert, Cornelia; Laccone, Franco (2006). "Williams-Beuren syndrome: Determination of deletion size using quantitative real-time PCR". International Journal of Molecular Medicine. 18 (5): 799–806. doi:10.3892/ijmm.18.5.799. PMID 17016608.
- Francke, U. (1999). "Williams-Beuren syndrome:genes and mechanisms". Human Molecular Genetics. 8 (10): 1947–54. doi:10.1093/hmg/8.10.1947. PMID 10469848.
- Tassabehji, M; Metcalfe, K; Karmiloff-Smith, A; Carette, MJ; Grant, J; Dennis, N; Reardon, W; Splitt, M; Read, AP; Donnai, D (January 1999). "Williams syndrome: use of chromosomal microdeletions as a tool to dissect cognitive and physical phenotypes". American Journal of Human Genetics. 64 (1): 118–25. doi:10.1086/302214. PMC 1377709. PMID 9915950.
- "Williams syndrome – Genetics Home Reference" Archived 2010-01-22 at the Wayback Machine. The U.S. National Library of Medicine. 2010.
- "Diagnosing Williams Syndrome". Guide to Williams Syndrome. Williams Syndrome Association. Archived from the original on 2011-12-16.
- Lowery, M. C.; Morris, C. A.; Ewart, A; Brothman, L. J.; Zhu, X. L.; Leonard, C. O.; Carey, J. C.; Keating, M; Brothman, A. R. (1995). "Strong correlation of elastin deletions, detected by FISH, with Williams syndrome: Evaluation of 235 patients". American Journal of Human Genetics. 57 (1): 49–53. PMC 1801249. PMID 7611295.
- Committee On, Genetics (2001). "American Academy of Pediatrics: Health care supervision for children with Williams syndrome". Pediatrics. 107 (5): 1192–204. PMID 11331709.
- Dykens, Elisabeth M.; Rosner, Beth A.; Ly, Tran; Sagun, Jaclyn (2005). "Music and Anxiety in Williams Syndrome: A Harmonious or Discordant Relationship?". American Journal on Mental Retardation. 110 (5): 346–58. doi:10.1352/0895-8017(2005)110[346:MAAIWS]2.0.CO;2. PMID 16080773.
- Morris, Colleen A. (1993-01-01). Pagon, Roberta A.; Adam, Margaret P.; Ardinger, Holly H.; Wallace, Stephanie E.; Amemiya, Anne; Bean, Lora JH; Bird, Thomas D.; Fong, Chin-To; Mefford, Heather C. (eds.). Williams Syndrome. Seattle (WA): University of Washington, Seattle. PMID 20301427. Archived from the original on 2017-01-18.
- Stromme, P.; Bjomstad, P. G.; Ramstad, K. (2002). "Prevalence Estimation of Williams Syndrome". Journal of Child Neurology. 17 (4): 269–71. doi:10.1177/088307380201700406. PMID 12088082.
- Morris, Colleen A. (2010). "Introduction: Williams syndrome". American Journal of Medical Genetics Part C. 154C (2): 203–8. doi:10.1002/ajmg.c.30266. PMC 2946897. PMID 20425781.
- Westfahl, Gary; Slusser, George Edgar (1999). Nursery Realms: Children in the Worlds of Science Fiction, Fantasy, and Horror. University of Georgia Press. p. 153. ISBN 9780820321448.
- "Gabrielle director achieves perfect pitch" Archived 9 September 2017 at the Wayback Machine The Georgia Straight, 24 January 2014.
- "About Jeremy". I Am Jeremy Vest. Archived from the original on 10 May 2016. Retrieved 15 May 2016.
Further reading
- Fung, Lawrence K.; Quintin, Eve-Marie; Haas, Brian W.; Reiss, Allan L. (2012). "Conceptualizing neurodevelopmental disorders through a mechanistic understanding of fragile X syndrome and Williams syndrome". Current Opinion in Neurology (Review). 25 (2): 112–24. doi:10.1097/WCO.0b013e328351823c. PMC 3680360. PMID 22395002.
- Karmiloff-Smith, A.; d'Souza, D.; Dekker, T. M.; Van Herwegen, J.; Xu, F.; Rodic, M.; Ansari, D. (2012). "Genetic and environmental vulnerabilities in children with neurodevelopmental disorders". Proceedings of the National Academy of Sciences (Review). 109: 17261–5. Bibcode:2012PNAS..109S7261K. doi:10.1073/pnas.1121087109. JSTOR 41763522. PMC 3477396. PMID 23045661.
- Kitagawa, Hirochika; Fujiki, Ryoji; Yoshimura, Kimihiro; Oya, Hiroyuki; Kato, Shigeaki (2011). "Williams syndrome is an epigenome-regulator disease". Endocrine Journal (Review). 58 (2): 77–85. doi:10.1507/endocrj.k10e-393. PMID 21242649.
- Musolino, Julien; Landau, Barbara (2012). "Genes, language, and the nature of scientific explanations: The case of Williams syndrome". Cognitive Neuropsychology (Review). 29 (1–2): 123–48. doi:10.1080/02643294.2012.702103. PMC 3478137. PMID 23017087.
External links
| Wikimedia Commons has media related to Williams syndrome. |
| Classification | |
|---|---|
| External resources |