VEGFR-2 inhibitor
VEGFR-2 inhibitor, also known as kinase insert domain receptor(KDR) inhibitor,[1] are tyrosine kinase receptor inhibitors that reduce angiogenesis or lymphangiogenesis, leading to anticancer activity. Generally they are small, synthesised molecules that bind competitively to the ATP-site of the tyrosine kinase domain. VEGFR-2 selective inhibitor can interrupt multiple signaling pathways involved in tumor, including proliferation, metastasis and angiogenesis.[2]
VEGFR
The VEGF receptor family contains three members, which are all receptor tyrosine kinases (VEGFR-1, VEGFR-2 and VEGFR-3).[3] VEGFR-1, or FLT-1 (fms-like tyrosine kinase), are important for hematopoietic stem cell development, VEGFR-2 (KDRor FLK-1 (fetal liver kinase)) are vital to [[vascular endothelial cell development and VEGFR-3 (FLT-4)[4] for lymphatic endothelial cell development.[2] Binding of VEGF to the VEGFR induces a conformational change in the receptor producing a signaling pathway.[3]
VEGFR-2
VEGFR-2 is a 210-230 kDa glycoprotein expressed in vascular endothelial cells and in hematopoietic stem cells and binds VEGF-A.[2][4] VEGFR-2 is closely related to VEGFR-1 for they have common and specific ligands but VEGFR-2 is a highly active kinase while VEGFR1 is an impaired receptor tyrosine kinase. This receptor is a regulator in responses in the endothelial cells of VEGF. These regulations include permeability, proliferation, invasion and migration. The signaling pathways, Y1175 and Y1214, are the main autophosphorylation sites of the human VEGFR-2 followed by VEGF binding. Autophosphorylated VEGFR-2 is required for activation of several downstream pathways, which are hyperactivated in some tumors.[2] These signaling pathways are important in tumor angiogenesis, which stimulates tumor growth by supplying the tumor with oxygen and nutrients.[3] VEGFR-2 is overexpressed in several cancers, for example ovarian, thyroid, melanoma and medulloblastoma.[2]
Structure
VEGFR-2 are a part of the VEGF family. The other receptors in the family are VEGFR-1 and VEGFR-3. These receptors are a type of transmembrane kinase receptors and have similar structure. They have an extracellular part that is made up of an N-terminus signal and a 7 immunoglobulin-like domain. The intracellular part of the receptor consists of a juxtamembrane (the tyrosine kinase domain), which is divided to proximal and distal kinase domains and a C-terminus tail.[2]
Medical Uses
Sorafenib is used as an oral drug which inhibits angiogenesis and tumor cell growth in VEGFR-2 and VEGFR-3 (inhibits the phosphorylation), PDGFRβ (platelet-derived growth factor receptor), RAF kinase, FLT3 (Fms-like tyrosine kinase-3) and KIT (stem cell factor receptor). Sorafenib is used in the treatment of advanced renal cell carcinoma. As of October 2018, it is in phase III of clinical trials for hepatocellular carcinoma, metastatic melanoma and non-small cell lung cancer (NSCLC).[5] Sunitinib is an oral drug that inhibits the phosphorylation of all the VEGF receptors, PDGFR-ß, KIT FLT3, CSF1R and GDNF. Sunitinib is used in the treatment of advanced renal cell carcinoma and gastrointestinal stromal tumors.[5] Bevacizumab is a humanized monoclonal antibody, which binds selectively to VEGF receptor. It is used as an injection therapy, often in combination with other drugs. It is used in combination with 5-fluorouracil as a first-line treatment of metastatic carcinoma in the colon or rectum. In advanced non-small cell lung cancer bevacizumab (Avastin) is used as a first-line treatment in combination with paclitaxel and carboplatin. It can also be used in the treatment for breast cancer and kidney cancer.[5] Lenvatinib and vandetanib are used for the treatment of progressive and locally advanced differentiated thyroid cancer (DTC).[6][7] Pazopanib and axitinib are used orally for the treatment of advanced renal cell carcinoma.[8][9] Cabozantinib has the same indication but it is only used on patients who have already received an anti-angiogenic therapy.[10] Regorafenib is administered orally for the treatment of colorectal cancer, gastrointestinal stromal cancer and hepatocellular carcinoma.[11] Nintedanib is used for the treatment of idiopathic pulmonary fibrosis. Tests for liver function have to be made to modify doses before the therapy can start.[12] Apatinib is used for the treatment of advanced gastric cancer. It is often diagnosed in lage stages because there are no early signs or symptoms.[13]
Mechanism of action
Binding to VEGF receptor induces dimerization, which modifies the conformation in the intracellular domain. This modification leads to the exposure of the ATP-binding site, which causes ATP binding on the receptor and also transphosphorylation on specific tyrosine residues. Tyrosine phosphorylation on the receptor is regulated by internalization, degradation and by dephosphorylation through different protein tyrosine phosphatases. This can then lead to the initial receptor signal transduction cascade, which activates several downstream enzymatic pathways.[2] The signaling inhibition of VEGFR is through tumor vessels and not the tumor cells. Reduction of VEGF expression reduces blood flow to tumor and stops tumor angiogenesis.[4]
Adverse Effects
VEGF inhibitors in the treatment of cancer often cause adverse effects. Treatment with VEGF inhibitors suppresses cellular signalling pathways that are important in microvasculature regulation and maintenance. The effects on normal organs can then lead to vascular disturbances and regression of blood vessels.[5]
FDA has approved three drugs, bevacizumab, sunitinib and sorafenib, that were developed for antiangiogenic actions and are used in the treatment of patients with specific cancer types. All of these drugs have the mechanism of inhibiting VEGF signalling by blocking either the function of the VEGF ligand or VEGF receptor.[5]
Bevacizumab is a function-blocking monoclonal antibody that binds selectively to VEGF. Generally it is well tolerated and safe but can have adverse effects which can be intensified by chemotherapeutic agents used at the same time. For bevacizumab the most common adverse effects are hypertension, epistaxis, proteinuria, upper respiratory infection, stomatitis, diarrhea or other symptoms from the gastrointestinal tract as well as dyspnea, fatigue and dermatitis. Serious adverse effects connected to bevacizumab are infrequent but among those listed are gastrointestinal perforation, arterial thromboembolic events, hypertensive crisis, neutropenia, complications with wound healing, haemorrhage, nephrotic syndrome, heart failure and reversible posterior leukoencephalopathy syndrome.[5]
Sunitinib is a small molecule inhibitor which inhibits phosphorylation of VEGF receptor among other receptors. Sunitinib is mostly well tolerated. Common adverse effects which have an incidence rate of 20% are fatigue, asthenia, diarrhea, nausea, dyspepsia, abdominal pain, constipation, hypertension, skin discoloration, altered taste, stomatitis and mild bleeding.[5]
Sorafenib is a small molecule inhibitor of many tyrosine kinase receptors such as VEGFR-2. Side effects are in most cases mild to moderate such as rash, hand-foot skin reaction, diarrhea and dermatitis, and occur in about 33-38% patients using sorafenib. Other side effects are mild hypertension, leukopenia and bleeding. Uncommon side effects are cardiac ischaemia or infarction, gastrointestinal perforation, life-threatening haemorrhage and reversible posterior leukoencephalopathy syndrome.[5]
Hypertension is one of the most common side effects regarding inhibition of VEGF signalling. VEGF increases synthesis of NO through upregulation of endothelial NO synthase and therefore inhibition of VEGF diminishes NO synthesis. Decrease in NO causes vasoconstriction, increased peripheral resistance and increased blood pressure. Hypertension caused by VEGF inhibition can usually be treated with oral antihypertensive agents.[5]
Proteinuria is common when VEGF signalling is inhibited which shows how important VEGF is for normal renal function. VEGFR-2 can be found on the glomerular capillary endothelial cells and is activated by VEGF. Proteinuria is in most cases asymptomatic and usually decreases when treatment ends.[5]
Impaired wound healing can be an adverse effect of VEGF inhibition as angiogenesis is an important step in wound healing.[5]
Gastrointestinal perforation can be caused by VEGF inhibition although the mechanism is unknown. Abscesses, diverticula as well as bowel resection and anastomosis have been related to some cases.[5]
Haemorrhage and thrombosis can occur when VEGF is inhibited as VEGF promotes endothelial cell survival and helps maintaining vascular integrity. When VEGF is inhibited, the regenerative capacity of endothelial cells may diminish and pro-coagulant phospholipids could be exposed on the plasma membrane or the underlying matrix, possibly leading to either thrombosis or haemorrhage. Since VEGF increases production of NO and prostacyclin, the inhibition of VEGF leads to decrease in both chemicals which contributes to thromboembolic events.[5]
Reversible posterior leukoencephalopathy is often attributed to hypertensive encephalopathy as well as endothelial dysfunction. This can cause focal cerebral oedema, vasospasms, and even a breakdown in the blood-brain barrier. Inhibition of VEGF is implicated as a factor in the pathophysiology of the disease but has not yet been replicated after VEGF inhibition in preclinical models.[5]
Endocrine dysfunction has been reported as an adverse effect of VEGF inhibition. Hyperthyroidism is one such, since thyroid function can be damaged by capillary regression around the follicles of the thyroid. The fenestrated capillaries of the pituitary, adrenal cortex and pancreatic isle have also been known to regress as an effect of VEGF inhibition. The thyroid-hypothalamic feedback loop can also be impaired due to VEGF inhibition, followed by raised TSH blood concentration.[5]
Interactions
Lenvatinib inhibits the liver enzyme CYP3A, which also happens to be a metabolic enzyme for the drug. It inhibits the UDP-glucuronosyltransferases UGT1A1 and UGT1A4. Lenvatinib induces CYP3A but not UGT1A1 and UGT1A4. Use of other drugs, especially ones metabolized by the liver enzyme CYP3A, should be monitored in case their plasma concentration changes. In vitro studies have shown that lenvatinib inhibits organic anion transporters 1 and 3 (OAT1 and OAT3).[6] Sunitinib is metabolized in the liver by CYP3A4. It interacts with inducers and inhibitors of CYP3A4 leading to a decrease or increase in the plasma concentration of particular drugs metabolized by the same pathway. It will not change the amount of drug metabolized by the enzyme, because it does not inhibit or induce the enzyme directly. Sunitinib is a substrate of P-glycoprotein and ABCG2 transporters. It acts as an inhibitor for both transporters, especially for ABCG2. Therefore, drugs that are substrates of these carriers will have modified pharmacokinetics.[14]
Plasma concentration of sorafenib and paclitaxel may be increased when the drugs are co-administered along with carboplatin. This has no effect on carboplatin. It also increases the AUC of docetaxel, doxorubicin and irinotecan but decreases the AUC of fluorouracil and neomycin, so it is cautionary to administer sorafenib with these drugs as it may alter the plasma concentration. Sorafenib is metabolized by CYP3A4 and UGT1A9. This means that drugs metabolized by these pathways have to be carefully administered. Sorafenib's inhibition of UGT1A9 and UGT1A1 may increase plasma concentration of other drugs. The same goes for the CYP2B6 and CYP2C8 pathways, they are inhibited by sorafenib. Giving sorafenib in combination with rifampicin or inducers of CYP3A4 can decrease plasma concentration of sorafenib. CYP3A4 inhibitors are unlikely to affect sorafenib. Sorafenib is a competitive inhibitor of CYP2C19, CYP2D6 and CYP3A4. It inhibits P-glycoprotein, therefore it can increase the plasma concentration of drugs which are P-glycoprotein substrates.[15] Pazopanib is metabolized in the liver by CYP3A4 enzyme. Strong CYP3A4 inhibitors, other than pazopanib, can increase the plasma concentration of pazopanib, and CYP3A4 inducers will do the opposite. Grapefruit juice is a CYP3A4 inhibitor and should be avoided when taking pazopanib. It is also a weak inhibitor of other liver enzymes, CYP2C8 and CYP2D6.[8] Axitinib is metabolized by CYP3A4 and UGT1A1. Strong inhibitors of CYP3A4 will increase the plasma concentration of axitinib, while weak inhibitors have less effect on the plasma concentration. Strong inducers of CYP3A4 will decrease the plasma concentration of axitinib and should be avoided.[16]
Pipeline drugs
Lucitanib is a tyrosine kinase activity inhibitor, highly selective for VEGFR types 1-3, FGFR types 1-2 and PDGFR alpha/beta.[17] Tumor types such as breast carcinoma show amplification of fibroblast growth factor related genes. Simultaneous inhibition of VEGF and FGF receptors in FGFR1 dependent tumors could be therapeutically advantageous.[18] Lucitanib has been shown to have promising efficacy, a manageable side-effect profile and clinical benefits in both FGF-aberrant and angiogenesis-sensitive populations leading to a phase II program being planned.[17] Motesanib is a small-molecule multikinase inhibitor highly selective for VEGFR 1-3, PDGFR and KIT. The drug has shown anti-tumor activity as a monotherapy in advanced solid tumors.[19] Vatalanib is an antiangiogenic VEGFR inhibiting molecule which is being researched as a potential treatment of solid tumors. Vatalanib inhibits VEGFR 1-4 although it has greater potency as an inhibitor of VEGFR 1-2. At concentrations under 10 μM, Vatalanib does not have cytotoxic or antiproliferative effects on cells that do not express VEGF. Specific inhibition of tumor-induced angiogenesis like the inhibition by Vatalanib can both prevent ongoing growth of tumors and the metastatic potential.[20] Cediranib is a multi VEGFR 1-3 inhibitor being tested as a maintenance treatment for patients with platinum sensitive relapsed ovarian cancer.[21][22] Cediranib stops blood flow to the site of the tumour and thereby inhibits its growth.[21]
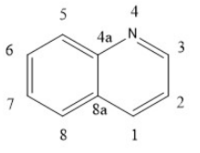
Structure-activity relationship (SAR)
Quinoline and quinazoline-derivatives
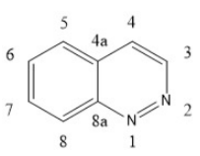
Lenvatinib, Vandetanib and Cabozantinib are drugs that belong to this group.[4] Novel biphenyl tricyclic quinazoline compounds and aryloxy quinolone derivatives are multiple kinase inhibitors. They are less likely to lead to drug resistance than selective inhibitors, which increases life expectancy. 4-quinazolinamine heterocyclic compounds and 2-chloro-4-anilino-quinazoline derivatives inhibit tumor vessel generation and restrain EGFR, HER-2, VEGFR-2 and mitosis process.[4] Quinoxaline derivatives with a diaryl-amide or diaryl-urea substructure have B-Raf mutant kinase inhibition activity. Some novel quinazoline derivatives inhibit Raf kinase selectively and have less effect on inhibition of VEGFR-2 and EGFR kinase. A scaffold in position N1 on quinolone and quinazoline derivatives behaves as a hydrogen bond receptor and interacts with Cys919 residue. The terminal substituent aromatic ring can form hydrophobic bonds with the hydrophobic pocket of VEGFR-2, especially the terminal phenyl group substituted by chloride in the para-position.[4] Quinolone-urea containing VEGFR inhibitors will bind to Asp1046 residue of the receptor via the carbonyl oxygen, and interact with Glu885 residue via two NH groups.[4]
Urea derivatives
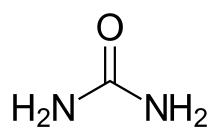
Urea derivatives on the market are Regorafenib and Sorafenib. The urea derivatives block the VEGFR and/or one or more protein kinases and can therefore modulate, regulate and/or inhibit tyrosine kinase signal transduction. Increased stability against degradation by liver enzymes can be acquired by adding a deuterium to heterocyclic compounds. Novel urea compounds with pentafluoro-sulfane substitute on a phenyl group show better protein kinase inhibition in diseases like cancer when compared to aryl-urea compounds with either quinazoline or pyrimidine moieties. N-substituted phenyl N’-substituted heterocyclic urea compounds give an IC50 between 15 nM and 1 μM for VEGFR-2. Having a 1H-indole-1-carboxamide scaffold on aryl-urea compounds results in added VEGFR-2 potency and selectivity and gives an IC50 of 3nM against the receptor.[4]
Indolin derivatives
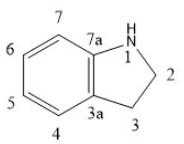
Indolin derivatives on the market are Sunitinib and Intedanib.[4]
Two indol derivatives that target the VEGF pathway, semaxanib and sunitinib, have been developed. The former is potent but was inefficient in clinical trials and the latter has many side effects. There is need for a drug with pharmacological effects similar to semaxanib and sunitinib, but it needs to be less toxic. MPEG3-9-semaxanib is semaxanib with an additional water-soluble, non-peptidic oligomer attached to it via a spacer moiety. MPEG3-9-semaxanib derivatives are 10 times more active against VEGFR-2 than sunitinib. Compounds with sunitinib heterocyclic moiety but different amide side chains inhibit VEGFR-1 and VEGFR-2 and regulate disorders. Another compound with sunitinib heterocyclic moiety and a pyrrole side chain has very good VEGFR-2 potency, with an IC50 of 65 nM. N-indol-1-amide compound is a possible anti-tumor drug in combination with other anticancer treatment and has an IC50 value of 31 nM.[4] There are many indol derivatives with different side chains that target multiple kinases and take part in several pathways in tumor development. Intedanib is a multiple tyrosine kinase inhibitor and is the first drug to treat idiopathic pulmonary fibrosis.[4] Indol derivatives with 1-NH of 2-indolinone motif that is an H-bond donor, and 2-carbonyl oxygen that acts as an H-bond acceptor, bind with Glu915 and Cys917, respectively. These compounds have basic amine side chains or nitrogen heterocycles and provide ideal solubility and pharmacokinetics. These compounds are promising anti-cancer and anti-fibrosis agents, inhibiting various protein tyrosine kinases. They have higher efficiency, lower toxicity, fewer side effects, favorable preparation technology and favorable physicochemical properties.[4]
Pyridine derivatives
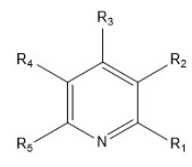
Pyridine derivatives on market are axitinib, regorafenib and apatinib.[4]
Pyridine derivatives with thiazolyamino-substituted heterocycle derivatives are kinase inhibitors. They show antiangiogenesis activity and anti-proliferation in tumor cells. The presence of sulfur atom in the heterocyclic ring is hypothesized to enhance potency on VEGFR-2 inhibition. Compounds with thieno [3,2-b] pyridine urea moiety are inhibitory for VEGF receptor signaling and HGF receptor signaling. HGF and HGF receptors undermine the activity of VEGF inhibition.[4] Pyridine derivatives with benzazepine have stronger selectivity and anti-tumor activity and less toxic side effects. They target many receptors like c-Met, VEGFR2, EGFR and therefore show inhibitory activity on a variety of tumors. Compounds with no fluorine atom in the benzazepine inhibit multiple targets and have better in vitro enzyme inhibiting activity.[4] 3-chloro derivatives and 3-methoxy-N-methyl-2-pyridine carboxamide derivatives inhibit, modulate and regulate tyrosine kinase signal transduction. VEGFR2 related diseases are treated with the compounds mentioned above in conjugation with other cancer therapies. 1-(5-tert-butyl-2-aryl-pyrazol-3-yl)-3-[2-fluoro-4-[(3-oxo-4H-pyrido[2,3-b] pyrazin-8-yl) oxy] phenyl] urea compounds are RAF inhibitors and treat disorders associated with mutated forms of RAF, like cancer, proliferative disorders, inflammation, immunological disorders, viral infections and fibrotic disorders.[4]
Pyrimidine derivatives
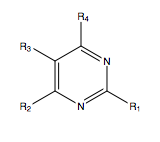
Pazopanib is a multi-targeted tyrosine kinase receptor inhibitor. The structure is formed by indazole, pyrimidine and a benzene ring.[23]
If the indazole ring is kept the same, replacing 5-amino-2-methylbenzenesulfonamide with another arylamine at position 2 of the pyrimidine, gives inhibitory effect on VEGFR-2 and c-Kit. For R2, two groups can show this effect on the compound, an ether group on one, and on the other a chloride group. When substituents are added on pazopanib with different electronic effects at the benzene ring the inhibition to the kinase changes.[23] Regarding the steric effect on position R1 and R2, a hydrogen in position R1 and a trifluoro-ether in R2 have the most inhibitory effect on VEGFR-2, which can be the effect of the electronegative groups. There is a steric hindrance of the indazole heterocycle, which can play an important role in the interaction with the tyrosine kinase receptor inhibition.[23]
Other small molecule VEGFR inhibitors
Conjugated 3-(indolyl)- and 3-(azaindolyl)-4-arylmaleimide compounds can induce apoptosis in cancer cells and therefore may have use in cancer therapy, including colorectal and gastric adenocarcinoma.[4]
Oxetane 3,3-dicarboxamide compounds are possible inhibitors of tumor angiogenesis and metastasis, and may also be effective against viral infections.[4] A 1,6-naphyridine-4-ketone fused heterocyclic derivative inhibits various kinases and the activity of tumor cells.[4] The antifungal drug itraconazole may be and inhibitor for VEGFR-2 and could be used in treatments that VEGFR-2 plays a role.[4] 3-phenyl-5-ureidoisothiazole-4-carboximide and 3-amino-5-phenylisothiazole derivatives inhibit, modulate and regulate tyrosine kinase signal and can be used for treatment of disorders that are caused by unregulated tyrosine kinase signal transduction, including cell growth, metabolic and blood vessel proliferative disorders.[4] Thioether derivatives can be used to treat all forms of cancer and target multi target protein kinase inhibitors.[4]
Pharmacology
Pharmacokinetics
Pharmacokinetics of Quinoline and quinazoline-derivatives
Absorption is variable within the group, with tmax ranging from 1–4 hours for Lenvatinib to 4–10 hours for cabozatinib. Food can slow down the absorption rate but should not affect the extent of the absorption. All drugs have high protein binding, ranging from 90-99%. Vandetanib and cabozatinib are metabolized mainly by CYP3A4, while lenvatinib is metabolized by CYP450 and via other pathways. The drugs are eliminated mostly in faeces, but also in urine. Vandetanib has a half-life of 19 days, while cabozatinib’s half-life is 99 hours.[24][25][26]
Pharmacokinetics of urea derivatives
Regorafenib and sorafenib reach mean peak plasma level in about 3 or 4 hours after a single oral dose. A high-fat meal decreases their absorption, while a low-fat meal may increase it, in comparison to taking the drugs in a fasting condition. In vitro protein binding is 99,5% for both drugs. The drugs are mainly metabolized in the liver by oxidative metabolism of CYP3A4, and glucuronidized by UGT1A9. Their half life ranges from 20 to 48 hours. Most of the administered dose should be out of the system in around 14 days. The drugs are mostly excreted in faeces, around 70-80%, but also in urine.[27][28]
Pharmacokinetics of indoline derivatives
Sunitinib's peak plasma level is reached 6–12 hours post-dose. The drug is 95% protein bound and has a volume of distribution of 2230 L, which indicates distribution into tissues. Sunitinib is metabolised mostly by CYP3A4 and has a half life of 40–60 hours.[29] Nintedanib reaches maximum plasma concentration 2–4 hours post-dose. The drug is 97,8% protein bound and is preferentially distributed in plasma. Only a minor part of the biotransformation of nintedanib is caused by CYP3A4 as the prevalent metabolic reaction for nintedanib is a hydrolytic cleavage of esterases. The half-life of nintedanib is about 10–15 hours.[30]
Pharmacokinetics of pyridine derivatives
Axitinib has short half-life, ranging from 2.5 to 6.1 hours, and therefore steady state should be reached in 2–3 days after the first dose. Peak plasma concentration is reached in 2.5 to 4.1 hours. In vitro protein binding is over 99%. Axitinib is primarily metabolized in the liver by CYP3A4/5. 30-60% of the drug is excreted in faeces, and about 23% in urine.[31] Regorafenib is a pyridine derivative, but also a urea derivative and has therefore been covered in that section.
Pharmacokinetics of pyrimidine derivatives
Pazopanib reaches maximum plasma concentration 3.5 hours post-dose. Pazopanib is about 99% protein bound to human plasma protein. Metabolism of pazopanib is mediated primarily by CYP3A4 and the half-life of the drug is about 30.9 hours. Elimination of pazopanib is primarily with faeces.[32]
History
Inhibition of angiogenesis including VEGFR-2 inhibitors has been of much interest and research in recent decades because angiogenesis is required for tumors to grow beyond a diameter of 1–2 mm. Many small molecular drugs and biological macromolecules targeting VEGFRs or blocking signal transduction of VEGF/VEGFR have been approved for clinical use or entered clinical trials.[4] In 2004 the monoclonal antibody bevacizumab became the first VEGFR inhibitor to be approved for cancer therapy.[33] The first small molecular VEGFR-2 inhibitor to be approved was sunitinib in 2006.[34]
References
- Harmange, Jean-Christophe; Weiss, Matthew M.; Germain, Julie; Polverino, Anthony J.; Borg, George; Bready, James; Chen, Danlin; Choquette, Deborah; Coxon, Angela; DeMelfi, Tom; DiPietro, Lucian; Doerr, Nicholas; Estrada, Juan; Flynn, Julie; Graceffa, Russell F.; Harriman, Shawn P.; Kaufman, Stephen; La, Daniel S.; Long, Alexander; Martin, Matthew W.; Neervannan, Sesha; Patel, Vinod F.; Potashman, Michele; Regal, Kelly; Roveto, Phillip M.; Schrag, Michael L.; Starnes, Charlie; Tasker, Andrew; Teffera, Yohannes; Wang, Ling; White, Ryan D.; Whittington, Douglas A.; Zanon, Roger (March 2008). "Naphthamides as Novel and Potent Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitors: Design, Synthesis, and Evaluation". Journal of Medicinal Chemistry. 51 (6): 1649–1667. doi:10.1021/jm701097z. ISSN 0022-2623. PMID 18324761.
- Musumeci, Francesca; Radi, Marco; Brullo, Chiara; Schenone, Silvia (25 October 2012). "Vascular Endothelial Growth Factor (VEGF) Receptors: Drugs and new inhibitors". Journal of Medicinal Chemistry. 55 (24): 10797–10822. doi:10.1021/jm301085w. PMID 23098265.
- Li, Chunpu; Ai, Jing; Zhang, Dengyou; Peng, Xia; Chen, Xi; Gao, Zhiwei; Su, Yi; Zhu, Wei; Ji, Yinchun; Chen, Xiaoyan; Geng, Meiyu; Liu, Hong (3 April 2015). "Design, Synthesis, and Biological Evaluation of Novel Imidazo[1,2-alpha]pyridine Derivatives as Potent c-Met Inhibitors". ACS Medicinal Chemistry Letters. 6 (5): 507–512. doi:10.1021/ml5004876. PMC 4434476. PMID 26005523.
- Peng, Fan-Wei; Liu, Da-Ke; Zhang, Qing-Wen; Xu, Yun-Gen; Shi, Lei (23 June 2017). "VEGFR-2 inhibitors and the therapeutic applications thereof: a patent review (2012-2016)". Expert Opinion on Therapeutic Patents. 27 (9): 987–1004. doi:10.1080/13543776.2017.1344215. PMID 28621580.
- Kamba, T; McDonald, D M (22 May 2007). "Mechanisms of adverse effects of anti-VEGF therapy for cancer". British Journal of Cancer. 96 (12): 1788–1795. doi:10.1038/sj.bjc.6603813. PMC 2359962. PMID 17519900.
- "LENVIMA" (PDF). FDA. Food and Drug Administration. Retrieved 20 September 2018.
- "CAPRELSA" (PDF). FDA. Food and Drug Administration. Retrieved 20 September 2018.
- "VOTRIENT" (PDF). FDA. Food and Drug Administration. Retrieved 20 September 2018.
- "INLYTA". Accessdata. Food and Drug Administration. Retrieved 20 September 2018.
- "CABOMETYX" (PDF). FDA. Food and Drug Administration. Retrieved 20 September 2018.
- "Stivarga (regorafenib) dosing, indications, interactions, adverse effects, and more". reference.medscape.com. Medscape. Retrieved 25 September 2018.
- "Vargatef" (PDF). www.ec.europa.eu. European medicines agency. Retrieved 27 September 2018.
- "The US FDA grants Apatinib Orphan Drug Designation for Treatment of Gastric Cancer – LSK BioPharma". lskbiopharma.com. LSK BioPharma. 19 June 2017. Retrieved 27 September 2018.
- Bilbao-Meseguer, Idoia; Jose, Begoña San; Lopez-Gimenez, Leocadio R; Gil, Maria A; Serrano, Laura; Castaño, Mikel; Sautua, Saioa; Basagoiti, Amaya De; Belaustegui, Ainhoa; Baza, Beatriz; Baskaran, Zuriñe; Bustinza, Alazne (8 January 2014). "Drug interactions with sunitinib". Journal of Oncology Pharmacy Practice. 21 (1): 52–66. doi:10.1177/1078155213516158. ISSN 1078-1552. PMID 24403097.
- "NEXAVAR" (PDF). FDA. Food and Drug Administration. Retrieved 20 September 2018.
- "INLYTA". Pfizer. Pfizer. Retrieved 20 September 2018.
- Soria, J.- C.; DeBraud, F.; Bahleda, R.; Adamo, B.; Andre, F.; Dientsmann, R.; Delmonte, A.; Cereda, R.; Isaacson, J.; Litten, J.; Allen, A.; Dubois, F.; Saba, C.; Robert, R.; D'Incalci, M.; Zucchetti, M.; Camboni, M. G.; Tabernero, J. (5 September 2014). "Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors". Annals of Oncology. 25 (11): 2244–2251. doi:10.1093/annonc/mdu390. ISSN 0923-7534. PMID 25193991.
- Guffanti, Federica; Chilà, Rosaria; Bello, Ezia; Zucchetti, Massimo; Zangarini, Monique; Ceriani, Laura; Ferrari, Mariella; Lupi, Monica; Jacquet-Bescond, Anne; Burbridge, Mike F.; Pierrat, Marie-Jeanne; Damia, Giovanna (15 December 2016). "In Vitro and In Vivo Activity of Lucitanib in FGFR1/2 Amplified or Mutated Cancer Models". Neoplasia (New York, N.Y.). 19 (1): 35–42. doi:10.1016/j.neo.2016.11.008. ISSN 1522-8002. PMC 5167242. PMID 27988457.
- Kubota, K.; Ichinose, Y.; Scagliotti, G.; Spigel, D.; Kim, J. H.; Shinkai, T.; Takeda, K.; Kim, S.- W.; Hsia, T.- C.; Li, R. K.; Tiangco, B. J.; Yau, S.; Lim, W.- T.; Yao, B.; Hei, Y.- J.; Park, K. (13 January 2014). "Phase III study (MONET1) of motesanib plus carboplatin/paclitaxel in patients with advanced nonsquamous nonsmall-cell lung cancer (NSCLC): Asian subgroup analysis". Annals of Oncology. 25 (2): 529–536. doi:10.1093/annonc/mdt552. ISSN 0923-7534. PMID 24419239.
- Jost, Lorenz M.; Gschwind, Hans-Peter; Jalava, Tarja; Wang, Yongyu; Guenther, Clemens; Souppart, Claire; Rottmann, Antje; Denner, Karsten; Waldmeier, Felix; Gross, Gerhard; Masson, Eric; Laurent, Dirk (1 November 2006). "Metabolism and Disposition of Vatalanib (PTK787/ZK-222584) in Cancer Patients". Drug Metabolism and Disposition. 34 (11): 1817–1828. doi:10.1124/dmd.106.009944. ISSN 0090-9556. PMID 16882767.
- "AstraZeneca provides update on cediranib EU marketing authorisation application". www.astrazeneca.com. AstraZeneca. Retrieved 28 September 2018.
- Nikolinakos, Petros; Heymach, John V (1 June 2008). "The Tyrosine Kinase Inhibitor Cediranib for Non-small Cell Lung Cancer and Other Thoracic Malignancies". Journal of Thoracic Oncology. 3 (6): S131–S134. doi:10.1097/JTO.0b013e318174e910. ISSN 1556-0864. PMID 18520296.
- Qi, Haofei; Ligong, Chen; Liu, Bingni; Wang, Xinran; Long, Li; Liu, Dengke (15 February 2014). "Synthesis and biological evaluation of novel pazopanib derivatives as antitumor agents". Bioorganic & Medicinal Chemistry Letters. 24 (4): 1108–1110. doi:10.1016/j.bmcl.2014.01.003. ISSN 0960-894X. PMID 24456902.
- "Lenvima" (PDF). www.ema.europa.eu. European medicines agency. Retrieved 28 September 2018.
- "Caprelsa" (PDF). www.ema.europa.eu. European medicines agency. Retrieved 28 September 2018.
- "CABOMETYX" (PDF). www.ema.europa.eu. European medicines agency. Retrieved 28 September 2018.
- "Stivarga" (PDF). www.ema.europa.eu. European medicines agency. Retrieved 28 September 2018.
- "Nexavar" (PDF). www.ema.europa.eu. European medicines agency. Retrieved 27 September 2018.
- "Sutent" (PDF). www.ema.europa.eu. European medicines agency. Retrieved 27 September 2018.
- "Vargatef" (PDF). www.ec.europa.eu. European medicines agency. Retrieved 27 September 2018.
- "Inlyta" (PDF). EMA. European medicines agency. Retrieved 27 September 2018.
- "Votrient" (PDF). www.ema.europa.eu. European medicines agency. Retrieved 26 September 2018.
- "Avastin approval history". www.drugs.com. Drugs.com. Retrieved 30 September 2018.
- "Sutent Approval History". www.drugs.com. Drugs.com. Retrieved 30 September 2018.