Rhizomelic chondrodysplasia punctata
Rhizomelic chondrodysplasia punctata is a rare developmental brain disorder characterized by systemic shortening of the proximal bones (i.e. rhizomelia), seizures, recurrent respiratory tract infections and congenital cataracts. The affected individuals have low levels of plasmalogens.[2]
| Rhizomelic chondrodysplasia punctata | |
|---|---|
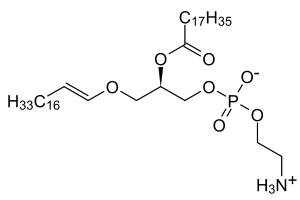 | |
| Low levels of plasmalogens is a characteristic of rhizomelic chondrodysplasia punctata. | |
| Specialty | Medical genetics |
| Symptoms | Alopecia, flat face[1] |
| Causes | PEX7 gene, GNPAT gene and AGPS gene mutations[2] |
| Diagnostic method | Clinical and radiologic finding[3] |
| Treatment | Physical therapy[4] |
Signs and symptoms
Rhizomelic chondrodysplasia punctata has the following symptoms:[4][1]
- Bilateral shortening of the femur
- Post-natal growth problems (deficiency)
- Cataracts
- Intellectual disability
- Possible seizures
- Possible infections of respiratory tract
Genetics
This condition is a consequence of mutations in the PEX7 gene, the GNPAT gene (which is located on chromosome 1) or the AGPS gene. The condition is acquired in an autosomal recessive manner.[2]
Pathophysiology
The mechanism of rhizomelic chondrodysplasia punctata in the case of type 1 of this condition one finds that peroxisome objective is PEX7, in peroxisome assembly. There are 3 pathways that count on PEX7 and are:[4][5]
- AGPS (catalyzes plasmalogen biosynthesis)
- PhYH (catalyzes catabolism of phytanic acid)
- ACAA1 (catalyzes beta-oxidation of VLCFA - straight)
Diagnosis
The diagnosis of rhizomelic chondrodysplasia punctata can be based on genetic testing[6] as well as radiography results, plus a physical examination of the individual.[3]
Treatment
Management of rhizomelic chondrodysplasia punctata can include physical therapy; additionally orthopedic procedures improved function sometimes in affected people.[4] However, the prognosis is poor in this condition.[3]
See also
References
- "Rhizomelic chondrodysplasia punctata type 1 | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program". rarediseases.info.nih.gov. Retrieved 23 January 2017.
- Reference, Genetics Home. "rhizomelic chondrodysplasia punctata". Genetics Home Reference. Retrieved 2017-01-16.
- RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: Rhizomelic chondrodysplasia punctata". www.orpha.net. Retrieved 23 January 2017.
- Braverman, Nancy E.; Moser, Ann B.; Steinberg, Steven J. (1 January 1993). "Rhizomelic Chondrodysplasia Punctata Type 1". GeneReviews. Retrieved 16 January 2017.update 2012
- Brodsky, Michael C. (2016-06-28). Pediatric Neuro-Ophthalmology. Springer. p. 620. ISBN 9781493933846.
- "Rhizomelic chondrodysplasia punctata type 1 - Conditions - GTR - NCBI". www.ncbi.nlm.nih.gov. Retrieved 23 January 2017.
- "OMIM Entry - # 222765 - RHIZOMELIC CHONDRODYSPLASIA PUNCTATA, TYPE 2; RCDP2". omim.org. Retrieved 16 January 2017.
- "OMIM Entry - # 600121 - RHIZOMELIC CHONDRODYSPLASIA PUNCTATA, TYPE 3; RCDP3". omim.org. Retrieved 2017-01-16.
Further reading
- Benacerraf, Beryl (2007). Ultrasound of fetal syndromes (2nd ed.). Philadelphia: Churchill Livingstone / Elsevier. ISBN 978-0443066412. Retrieved 23 January 2017.
- al.], [edited by] Kenneth F. Swaiman ... [et; Ashwal, Stephen; Ferriero, Donna M.; Schor, Nina F. (2012). Swaiman's pediatric neurology principles and practice (5th ed.). [Edinburgh]: Elsevier Saunders. ISBN 978-0323089111. Retrieved 23 January 2017.CS1 maint: extra text: authors list (link)
External links
| Classification | |
|---|---|
| External resources |