Non-coding DNA
Non-coding DNA sequences are components of an organism's DNA that do not encode protein sequences. Some non-coding DNA is transcribed into functional non-coding RNA molecules (e.g. transfer RNA, ribosomal RNA, and regulatory RNAs). Other functions of non-coding DNA include the transcriptional and translational regulation of protein-coding sequences, scaffold attachment regions, origins of DNA replication, centromeres and telomeres. Its RNA counterpart is non-coding RNA.
The amount of non-coding DNA varies greatly among species. Often, only a small percentage of the genome is responsible for coding proteins, but an increasing percentage is being shown to have regulatory functions. When there is much non-coding DNA, a large proportion appears to have no biological function, as predicted in the 1960s. Since that time, this non-functional portion has controversially been called "junk DNA".[1]
The international Encyclopedia of DNA Elements (ENCODE) project uncovered, by direct biochemical approaches, that at least 80% of human genomic DNA has biochemical activity.[2] Though this was not necessarily unexpected due to previous decades of research discovering many functional non-coding regions,[3][4] some scientists criticized the conclusion for conflating biochemical activity with biological function.[5][6][7][8][9] Estimates for the biologically functional fraction of the human genome based on comparative genomics range between 8 and 15%.[10][11][12] However, others have argued against relying solely on estimates from comparative genomics due to its limited scope. Non-coding DNA has been found to be involved in epigenetic activity and complex networks of genetic interactions and is being explored in evolutionary developmental biology.[4][11][13][14]
Fraction of non-coding genomic DNA

The amount of total genomic DNA varies widely between organisms, and the proportion of coding and non-coding DNA within these genomes varies greatly as well. For example, it was originally suggested that over 98% of the human genome does not encode protein sequences, including most sequences within introns and most intergenic DNA,[16] while 20% of a typical prokaryote genome is non-coding.[3]
In eukaryotes, genome size, and by extension the amount of non-coding DNA, is not correlated to organism complexity, an observation known as the C-value enigma.[17] For example, the genome of the unicellular Polychaos dubium (formerly known as Amoeba dubia) has been reported to contain more than 200 times the amount of DNA in humans.[18] The pufferfish Takifugu rubripes genome is only about one eighth the size of the human genome, yet seems to have a comparable number of genes; approximately 90% of the Takifugu genome is non-coding DNA.[16] Therefore, most of the difference in genome size is not due to variation in amount of coding DNA, rather, it is due to a difference in the amount of non-coding DNA.[19]
In 2013, a new "record" for the most efficient eukaryotic genome was discovered with Utricularia gibba, a bladderwort plant that has only 3% non-coding DNA and 97% of coding DNA. Parts of the non-coding DNA were being deleted by the plant and this suggested that non-coding DNA may not be as critical for plants, even though non-coding DNA is useful for humans.[15] Other studies on plants have discovered crucial functions in portions of non-coding DNA that were previously thought to be negligible and have added a new layer to the understanding of gene regulation.[20]
Types of non-coding DNA sequences
Cis- and trans-regulatory elements
Cis-regulatory elements are sequences that control the transcription of a nearby gene. Many such elements are involved in the evolution and control of development.[21] Cis-elements may be located in 5' or 3' untranslated regions or within introns. Trans-regulatory elements control the transcription of a distant gene.
Promoters facilitate the transcription of a particular gene and are typically upstream of the coding region. Enhancer sequences may also exert very distant effects on the transcription levels of genes.[22]
Introns
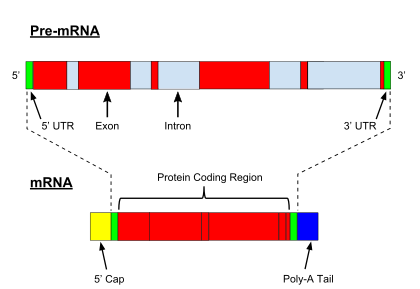
Introns are non-coding sections of a gene, transcribed into the precursor mRNA sequence, but ultimately removed by RNA splicing during the processing to mature messenger RNA. Many introns appear to be mobile genetic elements.[23]
Studies of group I introns from Tetrahymena protozoans indicate that some introns appear to be selfish genetic elements, neutral to the host because they remove themselves from flanking exons during RNA processing and do not produce an expression bias between alleles with and without the intron.[23] Some introns appear to have significant biological function, possibly through ribozyme functionality that may regulate tRNA and rRNA activity as well as protein-coding gene expression, evident in hosts that have become dependent on such introns over long periods of time; for example, the trnL-intron is found in all green plants and appears to have been vertically inherited for several billions of years, including more than a billion years within chloroplasts and an additional 2–3 billion years prior in the cyanobacterial ancestors of chloroplasts.[23]
Pseudogenes
Pseudogenes are DNA sequences, related to known genes, that have lost their protein-coding ability or are otherwise no longer expressed in the cell. Pseudogenes arise from retrotransposition or genomic duplication of functional genes, and become "genomic fossils" that are nonfunctional due to mutations that prevent the transcription of the gene, such as within the gene promoter region, or fatally alter the translation of the gene, such as premature stop codons or frameshifts.[24] Pseudogenes resulting from the retrotransposition of an RNA intermediate are known as processed pseudogenes; pseudogenes that arise from the genomic remains of duplicated genes or residues of inactivated genes are nonprocessed pseudogenes.[24] Transpositions of once functional mitochondrial genes from the cytoplasm to the nucleus, also known as NUMTs, also qualify as one type of common pseudogene.[25] Numts occur in many eukaryotic taxa.
While Dollo's Law suggests that the loss of function in pseudogenes is likely permanent, silenced genes may actually retain function for several million years and can be "reactivated" into protein-coding sequences[26] and a substantial number of pseudogenes are actively transcribed.[24][27] Because pseudogenes are presumed to change without evolutionary constraint, they can serve as a useful model of the type and frequencies of various spontaneous genetic mutations.[28]
Repeat sequences, transposons and viral elements
Transposons and retrotransposons are mobile genetic elements. Retrotransposon repeated sequences, which include long interspersed nuclear elements (LINEs) and short interspersed nuclear elements (SINEs), account for a large proportion of the genomic sequences in many species. Alu sequences, classified as a short interspersed nuclear element, are the most abundant mobile elements in the human genome. Some examples have been found of SINEs exerting transcriptional control of some protein-encoding genes.[29][30][31]
Endogenous retrovirus sequences are the product of reverse transcription of retrovirus genomes into the genomes of germ cells. Mutation within these retro-transcribed sequences can inactivate the viral genome.[32]
Over 8% of the human genome is made up of (mostly decayed) endogenous retrovirus sequences, as part of the over 42% fraction that is recognizably derived of retrotransposons, while another 3% can be identified to be the remains of DNA transposons. Much of the remaining half of the genome that is currently without an explained origin is expected to have found its origin in transposable elements that were active so long ago (> 200 million years) that random mutations have rendered them unrecognizable.[33] Genome size variation in at least two kinds of plants is mostly the result of retrotransposon sequences.[34][35]
Telomeres
Telomeres are regions of repetitive DNA at the end of a chromosome, which provide protection from chromosomal deterioration during DNA replication. Recent studies have shown that telomeres function to aid in its own stability. Telomeric repeat-containing RNA (TERRA) are transcripts derived from telomeres. TERRA has been shown to maintain telomerase activity and lengthen the ends of chromosomes.[36]
Junk DNA
The term "junk DNA" became popular in the 1960s.[37][38] According to T. Ryan Gregory, the nature of junk DNA was first discussed explicitly in 1972 by a genomic biologist, David Comings, who applied the term to all non-coding DNA.[39] The term was formalized that same year by Susumu Ohno,[19] who noted that the mutational load from deleterious mutations placed an upper limit on the number of functional loci that could be expected given a typical mutation rate. Ohno hypothesized that mammal genomes could not have more than 30,000 loci under selection before the "cost" from the mutational load would cause an inescapable decline in fitness, and eventually extinction. This prediction remains robust, with the human genome containing approximately (protein-coding) 20,000 genes. Another source for Ohno's theory was the observation that even closely related species can have widely (orders-of-magnitude) different genome sizes, which had been dubbed the C-value paradox in 1971.[6]
The term "junk DNA" has been questioned on the grounds that it provokes a strong a priori assumption of total non-functionality and some have recommended using more neutral terminology such as "non-coding DNA" instead.[39] Yet "junk DNA" remains a label for the portions of a genome sequence for which no discernible function has been identified and that through comparative genomics analysis appear under no functional constraint suggesting that the sequence itself has provided no adaptive advantage.
Since the late 70s it has become apparent that the majority of non-coding DNA in large genomes finds its origin in the selfish amplification of transposable elements, of which W. Ford Doolittle and Carmen Sapienza in 1980 wrote in the journal Nature: "When a given DNA, or class of DNAs, of unproven phenotypic function can be shown to have evolved a strategy (such as transposition) which ensures its genomic survival, then no other explanation for its existence is necessary."[40] The amount of junk DNA can be expected to depend on the rate of amplification of these elements and the rate at which non-functional DNA is lost.[41] In the same issue of Nature, Leslie Orgel and Francis Crick wrote that junk DNA has "little specificity and conveys little or no selective advantage to the organism".[42] The term occurs mainly in popular science and in a colloquial way in scientific publications, and it has been suggested that its connotations may have delayed interest in the biological functions of non-coding DNA.[43]
Some evidence indicate that some "junk DNA" sequences are sources for (future) functional activity in evolution through exaptation of originally selfish or non-functional DNA.[44]
ENCODE Project
In 2012, the ENCODE project, a research program supported by the National Human Genome Research Institute, reported that 76% of the human genome's non-coding DNA sequences were transcribed and that nearly half of the genome was in some way accessible to genetic regulatory proteins such as transcription factors.[1] However, the suggestion by ENCODE that over 80% of the human genome is biochemically functional has been criticized by other scientists,[5] who argue that neither accessibility of segments of the genome to transcription factors nor their transcription guarantees that those segments have biochemical function and that their transcription is selectively advantageous. After all, non-functional sections of the genome can be transcribed, given that transcription factors typically bind to short sequences that are found (randomly) all over the whole genome.[45]
Furthermore, the much lower estimates of functionality prior to ENCODE were based on genomic conservation estimates across mammalian lineages.[6][7][8][9] Wide-spread transcription and splicing in the human genome has been discussed as another indicator of genetic function in addition to genomic conservation which may miss poorly conserved functional sequences.[11] Furthermore, much of the apparent junk DNA is involved in epigenetic regulation and appears to be necessary for the development of complex organisms.[4][13][14] Genetic approaches may miss functional elements that do not manifest physically on the organism, evolutionary approaches have difficulties using accurate multispecies sequence alignments since genomes of even closely related species vary considerably, and with biochemical approaches, though having high reproducibility, the biochemical signatures do not always automatically signify a function.[11] Kellis et al. noted that 70% of the transcription coverage was less than 1 transcript per cell (and may thus be based on spurious background transcription). On the other hand, they argued that 12–15% fraction of human DNA may be under functional constraint, and may still be an underestimate when lineage-specific constraints are included. Ultimately genetic, evolutionary, and biochemical approaches can all be used in a complementary way to identify regions that may be functional in human biology and disease.[11] Some critics have argued that functionality can only be assessed in reference to an appropriate null hypothesis. In this case, the null hypothesis would be that these parts of the genome are non-functional and have properties, be it on the basis of conservation or biochemical activity, that would be expected of such regions based on our general understanding of molecular evolution and biochemistry. According to these critics, until a region in question has been shown to have additional features, beyond what is expected of the null hypothesis, it should provisionally be labelled as non-functional.[46]
Evidence of functionality
Some non-coding DNA sequences must have some important biological function. This is indicated by comparative genomics studies that report highly conserved regions of non-coding DNA, sometimes on time-scales of hundreds of millions of years. This implies that these non-coding regions are under strong evolutionary pressure and positive selection.[47] For example, in the genomes of humans and mice, which diverged from a common ancestor 65–75 million years ago, protein-coding DNA sequences account for only about 20% of conserved DNA, with the remaining 80% of conserved DNA represented in non-coding regions.[48] Linkage mapping often identifies chromosomal regions associated with a disease with no evidence of functional coding variants of genes within the region, suggesting that disease-causing genetic variants lie in the non-coding DNA.[48] The significance of non-coding DNA mutations in cancer was explored in April 2013.[49]
Non-coding genetic polymorphisms play a role in infectious disease susceptibility, such as hepatitis C.[50] Moreover, non-coding genetic polymorphisms contribute to susceptibility to Ewing sarcoma, an aggressive pediatric bone cancer.[51]
Some specific sequences of non-coding DNA may be features essential to chromosome structure, centromere function and recognition of homologous chromosomes during meiosis.[52]
According to a comparative study of over 300 prokaryotic and over 30 eukaryotic genomes,[53] eukaryotes appear to require a minimum amount of non-coding DNA. The amount can be predicted using a growth model for regulatory genetic networks, implying that it is required for regulatory purposes. In humans the predicted minimum is about 5% of the total genome.
Over 10% of 32 mammalian genomes may function through the formation of specific RNA secondary structures.[54] The study used comparative genomics to identify compensatory DNA mutations that maintain RNA base-pairings, a distinctive feature of RNA molecules. Over 80% of the genomic regions presenting evolutionary evidence of RNA structure conservation do not present strong DNA sequence conservation.
Non-coding DNA may perhaps serve to decrease the probability of gene disruption during chromosomal crossover.[55]
Regulating gene expression
Some non-coding DNA sequences determine the expression levels of various genes, both those that are transcribed to proteins and those that themselves are involved in gene regulation.[56][57][58]
Transcription factors
Some non-coding DNA sequences determine where transcription factors attach.[56] A transcription factor is a protein that binds to specific non-coding DNA sequences, thereby controlling the flow (or transcription) of genetic information from DNA to mRNA.[59][60]
Operators
An operator is a segment of DNA to which a repressor binds. A repressor is a DNA-binding protein that regulates the expression of one or more genes by binding to the operator and blocking the attachment of RNA polymerase to the promoter, thus preventing transcription of the genes. This blocking of expression is called repression.[61]
Enhancers
An enhancer is a short region of DNA that can be bound with proteins (trans-acting factors), much like a set of transcription factors, to enhance transcription levels of genes in a gene cluster.[62]
Silencers
A silencer is a region of DNA that inactivates gene expression when bound by a regulatory protein. It functions in a very similar way as enhancers, only differing in the inactivation of genes.[63]
Promoters
A promoter is a region of DNA that facilitates transcription of a particular gene when a transcription factor binds to it. Promoters are typically located near the genes they regulate and upstream of them.[64]
Insulators
A genetic insulator is a boundary element that plays two distinct roles in gene expression, either as an enhancer-blocking code, or rarely as a barrier against condensed chromatin. An insulator in a DNA sequence is comparable to a linguistic word divider such as a comma in a sentence, because the insulator indicates where an enhanced or repressed sequence ends.[65]
Uses
Evolution
Shared sequences of apparently non-functional DNA are a major line of evidence of common descent.[66]
Pseudogene sequences appear to accumulate mutations more rapidly than coding sequences due to a loss of selective pressure.[28] This allows for the creation of mutant alleles that incorporate new functions that may be favored by natural selection; thus, pseudogenes can serve as raw material for evolution and can be considered "protogenes".[67]
A study published in 2019 shows that new genes (termed de novo gene birth) can be fashioned from non-coding regions.[68] Some studies suggest at least one-tenth of genes could be made in this way.[68]
Long range correlations
A statistical distinction between coding and non-coding DNA sequences has been found. It has been observed that nucleotides in non-coding DNA sequences display long range power law correlations while coding sequences do not.[69][70][71]
Forensic anthropology
Police sometimes gather DNA as evidence for purposes of forensic identification. As described in Maryland v. King, a 2013 U.S. Supreme Court decision:[72]
The current standard for forensic DNA testing relies on an analysis of the chromosomes located within the nucleus of all human cells. 'The DNA material in chromosomes is composed of "coding" and "non-coding" regions. The coding regions are known as genes and contain the information necessary for a cell to make proteins. . . . Non-protein coding regions . . . are not related directly to making proteins, [and] have been referred to as "junk" DNA.' The adjective "junk" may mislead the lay person, for in fact this is the DNA region used with near certainty to identify a person.[72]
See also
References
- Pennisi E (September 2012). "Genomics. ENCODE project writes eulogy for junk DNA". Science. 337 (6099): 1159–1161. doi:10.1126/science.337.6099.1159. PMID 22955811.
- The ENCODE Project Consortium (September 2012). "An integrated encyclopedia of DNA elements in the human genome". Nature. 489 (7414): 57–74. Bibcode:2012Natur.489...57T. doi:10.1038/nature11247. PMC 3439153. PMID 22955616..
- Costa, Fabrico (2012). "7 Non-coding RNAs, Epigenomics, and Complexity in Human Cells". In Morris, Kevin V. (ed.). Non-coding RNAs and Epigenetic Regulation of Gene Expression: Drivers of Natural Selection. Caister Academic Press. ISBN 978-1904455943.
- Carey, Nessa (2015). Junk DNA: A Journey Through the Dark Matter of the Genome. Columbia University Press. ISBN 9780231170840.
- McKie, Robin (24 February 2013). "Scientists attacked over claim that 'junk DNA' is vital to life". The Observer.
- Eddy SR (November 2012). "The C-value paradox, junk DNA and ENCODE". Current Biology. 22 (21): R898–9. doi:10.1016/j.cub.2012.10.002. PMID 23137679.
- Doolittle WF (April 2013). "Is junk DNA bunk? A critique of ENCODE". Proceedings of the National Academy of Sciences of the United States of America. 110 (14): 5294–300. Bibcode:2013PNAS..110.5294D. doi:10.1073/pnas.1221376110. PMC 3619371. PMID 23479647.
- Palazzo AF, Gregory TR (May 2014). "The case for junk DNA". PLOS Genetics. 10 (5): e1004351. doi:10.1371/journal.pgen.1004351. PMC 4014423. PMID 24809441.
- Graur D, Zheng Y, Price N, Azevedo RB, Zufall RA, Elhaik E (2013). "On the immortality of television sets: "function" in the human genome according to the evolution-free gospel of ENCODE". Genome Biology and Evolution. 5 (3): 578–90. doi:10.1093/gbe/evt028. PMC 3622293. PMID 23431001.
- Ponting CP, Hardison RC (November 2011). "What fraction of the human genome is functional?". Genome Research. 21 (11): 1769–76. doi:10.1101/gr.116814.110. PMC 3205562. PMID 21875934.
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, et al. (April 2014). "Defining functional DNA elements in the human genome". Proceedings of the National Academy of Sciences of the United States of America. 111 (17): 6131–8. Bibcode:2014PNAS..111.6131K. doi:10.1073/pnas.1318948111. PMC 4035993. PMID 24753594.
- Rands CM, Meader S, Ponting CP, Lunter G (July 2014). "8.2% of the Human genome is constrained: variation in rates of turnover across functional element classes in the human lineage". PLOS Genetics. 10 (7): e1004525. doi:10.1371/journal.pgen.1004525. PMC 4109858. PMID 25057982.
- Mattick JS (2013). "The extent of functionality in the human genome". The HUGO Journal. 7 (1): 2. doi:10.1186/1877-6566-7-2. PMC 4685169.
- Morris K, ed. (2012). Non-Coding RNAs and Epigenetic Regulation of Gene Expression: Drivers of Natural Selection. Norfolk, UK: Caister Academic Press. ISBN 978-1904455943.
- "Worlds Record Breaking Plant: Deletes its Noncoding "Junk" DNA". Design & Trend. May 12, 2013. Retrieved 2013-06-04.
- Elgar G, Vavouri T (July 2008). "Tuning in to the signals: noncoding sequence conservation in vertebrate genomes". Trends in Genetics. 24 (7): 344–52. doi:10.1016/j.tig.2008.04.005. PMID 18514361.
- Thomas, C.A. (1971). "The genetic organization of chromosomes". Annu. Rev. Genet. 5: 237–256. doi:10.1146/annurev.ge.05.120171.001321. PMID 16097657.
- Gregory TR, Hebert PD (April 1999). "The modulation of DNA content: proximate causes and ultimate consequences". Genome Research. 9 (4): 317–24. doi:10.1101/gr.9.4.317 (inactive 2020-04-29). PMID 10207154.
- Ohno S (1972). Smith HH (ed.). "So Much "junk" DNA in Our Genome". Brookhaven Symposia in Biology. Gordon and Breach, New York. 23: 366–370. PMID 5065367. Retrieved 2013-05-15.
- Waterhouse PM, Hellens RP (April 2015). "Plant biology: Coding in non-coding RNAs". Nature. 520 (7545): 41–2. Bibcode:2015Natur.520...41W. doi:10.1038/nature14378. PMID 25807488.
- Carroll SB (July 2008). "Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution". Cell. 134 (1): 25–36. doi:10.1016/j.cell.2008.06.030. PMID 18614008.
- Visel A, Rubin EM, Pennacchio LA (September 2009). "Genomic views of distant-acting enhancers". Nature. 461 (7261): 199–205. Bibcode:2009Natur.461..199V. doi:10.1038/nature08451. PMC 2923221. PMID 19741700.
- Nielsen H, Johansen SD (2009). "Group I introns: Moving in new directions". RNA Biology. 6 (4): 375–83. doi:10.4161/rna.6.4.9334. PMID 19667762.
- Zheng D, Frankish A, Baertsch R, Kapranov P, Reymond A, Choo SW, Lu Y, Denoeud F, Antonarakis SE, Snyder M, Ruan Y, Wei CL, Gingeras TR, Guigó R, Harrow J, Gerstein MB (June 2007). "Pseudogenes in the ENCODE regions: consensus annotation, analysis of transcription, and evolution". Genome Research. 17 (6): 839–51. doi:10.1101/gr.5586307. PMC 1891343. PMID 17568002.
- Lopez JV, Yuhki N, Masuda R, Modi W, O'Brien SJ (1994). "Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat". Journal of Molecular Evolution. 39 (2): 174–190. doi:10.1007/bf00163806 (inactive 2020-04-29). PMID 7932781.
- Marshall CR, Raff EC, Raff RA (December 1994). "Dollo's law and the death and resurrection of genes". Proceedings of the National Academy of Sciences of the United States of America. 91 (25): 12283–7. Bibcode:1994PNAS...9112283M. doi:10.1073/pnas.91.25.12283. PMC 45421. PMID 7991619.
- Tutar Y (2012). "Pseudogenes". Comparative and Functional Genomics. 2012: 1–4. doi:10.1155/2012/424526. PMC 3352212. PMID 22611337.
- Petrov DA, Hartl DL (2000). "Pseudogene evolution and natural selection for a compact genome". The Journal of Heredity. 91 (3): 221–7. doi:10.1093/jhered/91.3.221. PMID 10833048.
- Ponicsan SL, Kugel JF, Goodrich JA (April 2010). "Genomic gems: SINE RNAs regulate mRNA production". Current Opinion in Genetics & Development. 20 (2): 149–55. doi:10.1016/j.gde.2010.01.004. PMC 2859989. PMID 20176473.
- Häsler J, Samuelsson T, Strub K (July 2007). "Useful 'junk': Alu RNAs in the human transcriptome". Cellular and Molecular Life Sciences (Submitted manuscript). 64 (14): 1793–800. doi:10.1007/s00018-007-7084-0. PMID 17514354.
- Walters RD, Kugel JF, Goodrich JA (August 2009). "InvAluable junk: the cellular impact and function of Alu and B2 RNAs". IUBMB Life. 61 (8): 831–7. doi:10.1002/iub.227. PMC 4049031. PMID 19621349.
- Nelson PN, Hooley P, Roden D, Davari Ejtehadi H, Rylance P, Warren P, Martin J, Murray PG (October 2004). "Human endogenous retroviruses: transposable elements with potential?". Clinical and Experimental Immunology. 138 (1): 1–9. doi:10.1111/j.1365-2249.2004.02592.x. PMC 1809191. PMID 15373898.
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. (February 2001). "Initial sequencing and analysis of the human genome". Nature. 409 (6822): 860–921. Bibcode:2001Natur.409..860L. doi:10.1038/35057062. PMID 11237011.
- Piegu B, Guyot R, Picault N, Roulin A, Sanyal A, Saniyal A, Kim H, Collura K, Brar DS, Jackson S, Wing RA, Panaud O (October 2006). "Doubling genome size without polyploidization: dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice". Genome Research. 16 (10): 1262–9. doi:10.1101/gr.5290206. PMC 1581435. PMID 16963705.
- Hawkins JS, Kim H, Nason JD, Wing RA, Wendel JF (October 2006). "Differential lineage-specific amplification of transposable elements is responsible for genome size variation in Gossypium". Genome Research. 16 (10): 1252–61. doi:10.1101/gr.5282906. PMC 1581434. PMID 16954538.
- Cusanelli E, Chartrand P (May 2014). "Telomeric noncoding RNA: telomeric repeat-containing RNA in telomere biology". Wiley Interdisciplinary Reviews: RNA. 5 (3): 407–19. doi:10.1002/wrna.1220. PMID 24523222.
- Ehret CF, De Haller G (October 1963). "Origin, development, and maturation of organelles and organelle systems of the cell surface in Paramecium". Journal of Ultrastructure Research. 23: SUPPL6:1–42. doi:10.1016/S0022-5320(63)80088-X. PMID 14073743.
- Dan Graur, The Origin of Junk DNA: A Historical Whodunnit
- Gregory, T. Ryan, ed. (2005). The Evolution of the Genome. Elsevier. pp. 29–31. ISBN 978-0123014634.
Comings (1972), on the other hand, gave what must be considered the first explicit discussion of the nature of "junk DNA," and was the first to apply the term to all non-coding DNA."; "For this reason, it is unlikely that any one function for non-coding DNA can account for either its sheer mass or its unequal distribution among taxa. However, dismissing it as no more than "junk" in the pejorative sense of "useless" or "wasteful" does little to advance the understanding of genome evolution. For this reason, the far less loaded term "noncoding DNA" is used throughout this chapter and is recommended in preference to "junk DNA" for future treatments of the subject."
- Doolittle WF, Sapienza C (April 1980). "Selfish genes, the phenotype paradigm and genome evolution". Nature. 284 (5757): 601–3. Bibcode:1980Natur.284..601D. doi:10.1038/284601a0. PMID 6245369.
- Another source is genome duplication followed by a loss of function due to redundancy.
- Orgel LE, Crick FH (April 1980). "Selfish DNA: the ultimate parasite". Nature. 284 (5757): 604–7. Bibcode:1980Natur.284..604O. doi:10.1038/284604a0. PMID 7366731.
- Khajavinia A, Makalowski W (May 2007). "What is "junk" DNA, and what is it worth?". Scientific American. 296 (5): 104. Bibcode:2007SciAm.296c.104.. doi:10.1038/scientificamerican0307-104. PMID 17503549.
The term "junk DNA" repelled mainstream researchers from studying noncoding genetic material for many years
- Biémont C, Vieira C (October 2006). "Genetics: junk DNA as an evolutionary force". Nature. 443 (7111): 521–4. Bibcode:2006Natur.443..521B. doi:10.1038/443521a. PMID 17024082.
- Lambert, Samuel A.; Jolma, Arttu; Campitelli, Laura F.; Das, Pratyush K.; Yin, Yimeng; Albu, Mihai; Chen, Xiaoting; Taipale, Jussi; Hughes, Timothy R.; Weirauch, Matthew T. (02 08, 2018). "The Human Transcription Factors". Cell. 172 (4): 650–665. doi:10.1016/j.cell.2018.01.029. ISSN 1097-4172. PMID 29425488. Check date values in:
|date=(help) - Palazzo AF, Lee ES (2015). "Non-coding RNA: what is functional and what is junk?". Frontiers in Genetics. 6: 2. doi:10.3389/fgene.2015.00002. PMC 4306305. PMID 25674102.
- Ludwig MZ (December 2002). "Functional evolution of noncoding DNA". Current Opinion in Genetics & Development. 12 (6): 634–9. doi:10.1016/S0959-437X(02)00355-6. PMID 12433575.
- Cobb J, Büsst C, Petrou S, Harrap S, Ellis J (April 2008). "Searching for functional genetic variants in non-coding DNA". Clinical and Experimental Pharmacology & Physiology. 35 (4): 372–5. doi:10.1111/j.1440-1681.2008.04880.x. PMID 18307723.
- Khurana E, Fu Y, Colonna V, Mu XJ, Kang HM, Lappalainen T, et al. (October 2013). "Integrative annotation of variants from 1092 humans: application to cancer genomics". Science. 342 (6154): 1235587. doi:10.1126/science.1235587. hdl:11858/00-001M-0000-0019-02F5-1. PMC 3947637. PMID 24092746.
- Lu YF, Mauger DM, Goldstein DB, Urban TJ, Weeks KM, Bradrick SS (November 2015). "IFNL3 mRNA structure is remodeled by a functional non-coding polymorphism associated with hepatitis C virus clearance". Scientific Reports. 5: 16037. Bibcode:2015NatSR...516037L. doi:10.1038/srep16037. PMC 4631997. PMID 26531896.
- Grünewald TG, Bernard V, Gilardi-Hebenstreit P, Raynal V, Surdez D, Aynaud MM, Mirabeau O, Cidre-Aranaz F, Tirode F, Zaidi S, Perot G, Jonker AH, Lucchesi C, Le Deley MC, Oberlin O, Marec-Bérard P, Véron AS, Reynaud S, Lapouble E, Boeva V, Rio Frio T, Alonso J, Bhatia S, Pierron G, Cancel-Tassin G, Cussenot O, Cox DG, Morton LM, Machiela MJ, Chanock SJ, Charnay P, Delattre O (September 2015). "Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite". Nature Genetics. 47 (9): 1073–8. doi:10.1038/ng.3363. PMC 4591073. PMID 26214589.
- Subirana JA, Messeguer X (March 2010). "The most frequent short sequences in non-coding DNA". Nucleic Acids Research. 38 (4): 1172–81. doi:10.1093/nar/gkp1094. PMC 2831315. PMID 19966278.
- Ahnert SE, Fink TM, Zinovyev A (June 2008). "How much non-coding DNA do eukaryotes require?". Journal of Theoretical Biology. 252 (4): 587–92. arXiv:q-bio/0611047. doi:10.1016/j.jtbi.2008.02.005. PMID 18384817.
- Smith MA, Gesell T, Stadler PF, Mattick JS (September 2013). "Widespread purifying selection on RNA structure in mammals". Nucleic Acids Research. 41 (17): 8220–36. doi:10.1093/nar/gkt596. PMC 3783177. PMID 23847102.
- Dileep, V. (2009). "The place and function of non-coding DNA in the evolution of variability". Hypothesis. 7 (1): e7. doi:10.5779/hypothesis.v7i1.146.
- Callaway, Ewen (March 2010). "Junk DNA gets credit for making us who we are". New Scientist.
- Carroll SB, Prud'homme B, Gompel N (May 2008). "Regulating evolution". Scientific American. 298 (5): 60–7. Bibcode:2008SciAm.298e..60C. doi:10.1038/scientificamerican0508-60. PMID 18444326.
- Stojic L, Niemczyk M, Orjalo A, Ito Y, Ruijter AE, Uribe-Lewis S, Joseph N, Weston S, Menon S, Odom DT, Rinn J, Gergely F, Murrell A (February 2016). "Transcriptional silencing of long noncoding RNA GNG12-AS1 uncouples its transcriptional and product-related functions". Nature Communications. 7: 10406. Bibcode:2016NatCo...710406S. doi:10.1038/ncomms10406. PMC 4740813. PMID 26832224.
- Latchman DS (December 1997). "Transcription factors: an overview". The International Journal of Biochemistry & Cell Biology. 29 (12): 1305–12. doi:10.1016/S1357-2725(97)00085-X. PMC 2002184. PMID 9570129.
- Karin M (February 1990). "Too many transcription factors: positive and negative interactions". The New Biologist. 2 (2): 126–31. PMID 2128034.
- Lewin, Benjamin (1990). Genes IV (4th ed.). Oxford: Oxford University Press. pp. 243–58. ISBN 978-0-19-854267-4.
- Blackwood EM, Kadonaga JT (July 1998). "Going the distance: a current view of enhancer action". Science. 281 (5373): 60–3. Bibcode:1998Sci...281...60.. doi:10.1126/science.281.5373.60. PMID 9679020.
- Maston GA, Evans SK, Green MR (2006). "Transcriptional regulatory elements in the human genome". Annual Review of Genomics and Human Genetics. 7: 29–59. doi:10.1146/annurev.genom.7.080505.115623. PMID 16719718.
- "Analysis of Biological Networks: Transcriptional Networks – Promoter Sequence Analysis" (PDF). Tel Aviv University. Retrieved 30 December 2012.
- Burgess-Beusse B, Farrell C, Gaszner M, Litt M, Mutskov V, Recillas-Targa F, Simpson M, West A, Felsenfeld G (December 2002). "The insulation of genes from external enhancers and silencing chromatin". Proceedings of the National Academy of Sciences of the United States of America. 99 Suppl 4: 16433–7. Bibcode:2002PNAS...9916433B. doi:10.1073/pnas.162342499. PMC 139905. PMID 12154228.
- "Plagiarized Errors and Molecular Genetics", talkorigins, by Edward E. Max, M.D., Ph.D.
- Balakirev ES, Ayala FJ (2003). "Pseudogenes: are they "junk" or functional DNA?". Annual Review of Genetics. 37: 123–51. doi:10.1146/annurev.genet.37.040103.103949. PMID 14616058.
- Levy, Adam (16 October 2019). "How evolution builds genes from scratch - Scientists long assumed that new genes appear when evolution tinkers with old ones. It turns out that natural selection is much more creative". Nature. 574 (7778): 314–316. doi:10.1038/d41586-019-03061-x. PMID 31619796.
- Peng CK, Buldyrev SV, Goldberger AL, Havlin S, Sciortino F, Simons M, Stanley HE (March 1992). "Long-range correlations in nucleotide sequences". Nature. 356 (6365): 168–70. Bibcode:1992Natur.356..168P. doi:10.1038/356168a0. PMID 1301010.
- Li W, Kaneko K (1992). "Long-Range Correlation and Partial 1/falpha Spectrum in a Non-Coding DNA Sequence" (PDF). Europhys. Lett. 17 (7): 655–660. Bibcode:1992EL.....17..655L. CiteSeerX 10.1.1.590.5920. doi:10.1209/0295-5075/17/7/014.
- Buldyrev SV, Goldberger AL, Havlin S, Mantegna RN, Matsa ME, Peng CK, Simons M, Stanley HE (May 1995). "Long-range correlation properties of coding and noncoding DNA sequences: GenBank analysis". Physical Review E. 51 (5): 5084–91. Bibcode:1995PhRvE..51.5084B. doi:10.1103/PhysRevE.51.5084. PMID 9963221.
- Slip opinion for Maryland v. King from the U.S. Supreme Court. Retrieved 2013-06-04.
Further reading
- Bennett, Michael D.; Leitch, Ilia J. (2005). "Genome size evolution in plants". In Gregory, T. Ryan (ed.). The Evolution of the Genome. San Diego: Elsevier. pp. 89–162. ISBN 978-0-08-047052-8.
- Gregory, T.R (2005). "Genome size evolution in animals". In T.R. Gregory (ed.). The Evolution of the Genome. San Diego: Elsevier. ISBN 978-0-12-301463-4.
- Shabalina SA, Spiridonov NA (2004). "The mammalian transcriptome and the function of non-coding DNA sequences". Genome Biology. 5 (4): 105. doi:10.1186/gb-2004-5-4-105. PMC 395773. PMID 15059247.
- Castillo-Davis CI (October 2005). "The evolution of noncoding DNA: how much junk, how much func?". Trends in Genetics. 21 (10): 533–6. doi:10.1016/j.tig.2005.08.001. PMID 16098630.