Phylogenetic footprinting
Phylogenetic footprinting is a technique used to identify transcription factor binding sites (TFBS) within a non-coding region of DNA of interest by comparing it to the orthologous sequence in different species. When this technique is used with a large number of closely related species, this is called phylogenetic shadowing.[1]
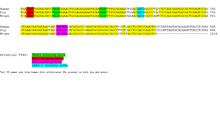
Researchers have found that non-coding pieces of DNA contain binding sites for regulatory proteins that govern the spatiotemporal expression of genes. These transcription factor binding sites (TFBS), or regulatory motifs, have proven hard to identify, primarily because they are short in length, and can show sequence variation. The importance of understanding transcriptional regulation to many fields of biology has led researchers to develop strategies for predicting the presence of TFBS, many of which have led to publicly available databases. One such technique is Phylogenetic Footprinting.
Phylogenetic footprinting relies upon two major concepts:
- The function and DNA binding preferences of transcription factors are well-conserved between diverse species.
- Important non-coding DNA sequences that are essential for regulating gene expression will show differential selective pressure. A slower rate of change occurs in TFBS than in other, less critical, parts of the non-coding genome.[2]
History
Phylogenetic footprinting was first used and published by Tagle et al. in 1988, which allowed researchers to predict evolutionary conserved cis-regulatory elements responsible for embryonic ε and γ globulin gene expression in primates.[3]
Before phylogenetic footprinting, DNase footprinting was used, where protein would be bound to DNA transcription factor binding sites (TFBS) protecting it from DNase digestion. One of the problems with this technique was the amount of time and labor it would take. Unlike DNase footprinting, phylogenetic footprinting relies on evolutionary constraints within the genome, with the "important" parts of the sequence being conserved among the different species.[4]
Protocol

It is important when using this technique to decide which genome your sequence should be aligned to. More divergent species will have less sequence similarity between orthologous genes. Therefore, the key is to pick species that are related enough to detect homology, but divergent enough to maximize non-alignment "noise". Step wise approach to Phylogenetic footprinting consists of :
- One should decide on the gene of interest.
- Carefully choose species with orthologous genes.
- Decide on the length of the upstream or maybe downstream region to be looked at.
- Align the sequences.
- Look for conserved regions and analyse them.
Not all TFBS are found
Not all transcription binding sites can be found using phylogenetic footprinting due to the statistical nature of this technique. Here are several reasons why some TFBS are not found:
Species specific binding sites
Some binding sites seem to have no significant matches in most other species. Therefore, detecting these sites by phylogenetic footprinting is likely impossible unless a large number of closely related species are available.
Very short binding sites
Some binding sites show excellent conservation, but just in a shorter region than the ones were looked for. Such short motifs (e.g., GC-box) often occur by chance in nonfunctional sequences and detecting these motifs can be challenging.
Less specific binding factors
Some binding sites show some conservation but have had insertions or deletions. It is not obvious if these sequences with insertions or deletions are still functional. Though they may still be functional if the binding factor is less specific (or less 'picky' if you will). Because deletions and insertions are rare in binding sites, considering insertions and deletions in the sequence would detect a few more true TFBSs, but it could likely include many more false positives.
Not enough data
Some motifs are quite well conserved, but they are statistically insignificant in a specific dataset. The motif might have appeared in different species by chance. These motifs could be detected if sequences from more organisms are available. So this will be less of a problem in the future.
Compound binding regions
Some transcription factors bind as dimers. Therefore, their binding sites may consist of two conserved regions, separated by a few variable nucleotides. Because of the variable internal sequence, the motif cannot be detected. However, if we could use a program to search for motifs containing a variable sequence in the middle, without counting mutations, these motifs could be discovered.
Accuracy
It is important to keep in mind that not all conserved sequences are under selection pressure. To eliminate false positives statistical analysis must be performed that will show that the motifs reported have a mutation rate meaningfully less than that of the surrounding nonfunctional sequence.
Moreover, results could be more accurate if the prior knowledge about the sequence is considered. For example, some regulatory elements are repeated 15 times in a promoter region (e.g., some metallothionein promoters have up to 15 metal response elements (MREs)). Thus, to eliminate false motifs with inconsistent order across species, the orientation and order of regulatory elements in a promoter region should be the same in all species. This type of information could help us to identify regulatory elements that are not adequately conserved but occur in several copies in the input sequence.[5]
References
- Phylogenetic Shadowing of Primate Sequences to Find Functional Regions of the Human Genome doi:10.1126/science.1081331
- Neph, S. and Tompa, M. 2006. MicroFootPrinter: a tool for phylogenetic footprinting in prokaryotic genomes. Nucleic Acids Research. 34: 366-368
- Tagle, D. A., Koop, B. F., Goodman, M., Slightom, J. L., Hess, D., and Jones, R. T. 1988. Embryonic ε and γ globin genes of a prosimian primate (Galago crassicaudatis): nucleotide and amino acid sequences, developmental regulation, and phylogenetic footprints. J. Mol. Biol. 203:439-455.
- Zhang, Z. and Gerstein, M. 2003. Of mice and men: phylogenetic footprinting aids the discovery of regulatory elements.J. Biol.2:11-11.4
- Blanchette, M. and Tompa, M. 2002. Discovery of Regulatory Elements by a Computational Method for Phylogenetic Footprinting. Genome Res. 12: 739-748