Laminopathy
Laminopathies (lamino- + -opathy) are a group of rare genetic disorders caused by mutations in genes encoding proteins of the nuclear lamina. They are included in the more generic term nuclear envelopathies that was coined in 2000 for diseases associated with defects of the nuclear envelope.[2] Since the first reports of laminopathies in the late 1990s, increased research efforts have started to uncover the vital role of nuclear envelope proteins in cell and tissue integrity in animals.
| Laminopathy | |
|---|---|
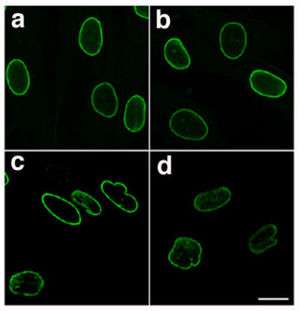 | |
| Normal nuclear lamina (a and b) and mutant nuclear lamina (c and d) from a patient with HGPS, visualized by immunofluorescence - note the irregular and bumpy shape of the laminopathic nuclei[1] | |
| Specialty | Clinical Genetics |
| Symptoms | Muscle weakness, reduced sensation, shortness of breath, syncope |
| Complications | Diabetes, heart failure, arrhythmias |
| Usual onset | Variable |
| Duration | Lifelong |
| Causes | Genetic |
| Diagnostic method | Clinical, genetic testing |
| Treatment | Physiotherapy, orthopaedic surgery, pacemaker, implantable defibrillator |
| Medication | ACE inhibitor, beta blocker, aldosterone antagonist |
| Prognosis | Variable |
Symptoms
Laminopathies and other nuclear envelopathies have a large variety of clinical symptoms including skeletal and/or cardiac muscular dystrophy, lipodystrophy and diabetes, dysplasia, dermo- or neuropathy, leukodystrophy, and progeria (premature aging). Most of these symptoms develop after birth, typically during childhood or adolescence. Some laminopathies however may lead to an early death, and mutations of lamin B1 (LMNB1 gene) may be lethal before or at birth.[3]
Genetics
Patients with classical laminopathy have mutations in the gene coding for lamin A/C (LMNA gene).
Mutations in the gene coding for lamin B2 (LMNB2 gene) have been linked to Barraquer-Simons syndrome[4] and duplication in the gene coding for lamin B1 (LMNB1 gene) cause autosomal dominant leukodystrophy.[5]
Mutations implicated in other nuclear envelopathies were found in genes coding for lamin-binding proteins such as lamin B receptor (LBR gene), emerin (EMD gene) and LEM domain-containing protein 3 (LEMD3 gene) and prelamin A-processing enzymes such as the zinc metalloproteinase STE24 (ZMPSTE24 gene).
Mutations causing laminopathies include recessive as well as dominant alleles with rare de novo mutations creating dominant alleles that do not allow their carriers to reproduce before death.
The nuclear envelopathy with the highest frequency in human populations is Emery–Dreifuss muscular dystrophy caused by an X-linked mutation in the EMD gene coding for emerin and affecting an estimated 1 in 100,000 people.
Molecular action
Lamins are intermediate filament proteins that form the nuclear lamina scaffold underneath the nuclear envelope in animal cells. They are attached to the nuclear envelope membrane via farnesyl anchors and interaction with inner nuclear membrane proteins such as lamin B receptor and emerin. The nuclear lamina appears to be an adaptation to mobility in animals as sessile organisms such as plants or fungi do not have lamins[6] and the symptoms of many laminopathies include muscle defects. Mutations in these genes might lead to defects in filament assembly and/or attachment to the nuclear envelope and thus jeopardize nuclear envelope stability in physically stressed tissues such as muscle fibers, bone, skin and connective tissue.[7]
Messenger RNA produced from the LMNA gene undergoes alternative splicing and is translated into lamins A and C. Lamin A undergoes farnesylation to attach a membrane anchor to the protein. This version of the protein is also referred to as prelamin A. Farnesylated prelamin A is further processed into mature lamin A by a metalloproteinase removing the last 15 amino acids and its farnesylated cysteine. This allows lamin A to dissociate from the nuclear envelope membrane and fulfill nuclear functions. Mutations causing laminopathies interfere with these processes on different levels.
Nonsense and missense mutations
Missense mutations in the lamin A/C rod and tail domains are the cause for a wide array of genetic disorders, suggesting that lamin A/C protein contains distinct functional domains that are essential for the maintenance and integrity of different cell lineages. Interaction between lamin A and the nuclear envelope protein emerin appears to be crucial in muscle cells, with certain mutations in lamin mimicking mutations in emerin and causing Emery–Dreifuss muscular dystrophy. Different mutations lead to dominant-negative and recessive alleles. Mutations in the lamin rod domain leading to mislocalization of both lamin A and emerin occur in patients with autosomal dominant forms of muscular dystrophy and cardiomyopathy.
Most lamin B mutations appear to be lethal with mutations in lamin B1 causing death at birth in mice.[3] In 2006, lamin B2 missense mutations were identified in patients with acquired partial lipodystrophy.[8]
Point mutations
The most common mutation in the lamin A/C is the homozygous Arg527His (arginine replaced by histidine at position 527) substitution in exon 9 of the LMNA gene[9] Other known mutations are Ala529Val and Arg527His/Val440Met.[10] Additionally, some mutations such as Arg527Cys, Lys542Asn, Arg471Cys, Thr528Met/Met540Thr, and Arg471Cys/Arg527Cys, Arg527Leu result in mandibuloacral dysplasia with progeria-like features.[11]
Splicing defects
Mutations causing progeria are defective in splicing LMNA mRNA, therefore producing abnormal lamin A protein, also known as progerin. The mutations activate a cryptic splice site within exon 11 of the gene, thereby causing the deletion of the processing site on prelamin A.[12] This results in an accumulation of progerin that is unable to mature into lamin A, leading to misshapen nuclei. Missplicing also leads to the complete or partial loss of exon 11 and results in a truncated prelamin A protein in the neonatal lethal tight skin contracture syndrome.
Processing defects
Since the metalloproteinase STE24 is required to process prelamin A into mature lamin A, mutations in this gene abolishing protease activity cause defects similar to laminopathies caused by prelamin A with truncated processing sites. Symptoms in patients with ZMPSTE24 mutation range from mandibuloacral dysplasia, progeroid appearance, and generalized lipodystrophy to infant-lethal restrictive dermopathy.
Gene dosage effects
In the case of autosomal dominant leukodystrophy, the disease is associated with a duplication of the lamin B gene LMNB1. The exact dosage of lamin B in cells appears to be crucial for nuclear integrity as increased expression of lamin B causes a degenerative phenotype in fruit flies and leads to abnormal nuclear morphology.[14]
Autoimmune antibodies
Antibodies against lamins are detected in the sera of some individuals with autoimmune diseases.[15]
DNA repair
A-type lamins promote genetic stability by maintaining the levels of proteins that have key roles in DNA double-strand break repair during the processes of non-homologous end joining and homologous recombination.[16] Mutations in lamin A (LMNA) cause Hutchinson–Gilford progeria syndrome, a dramatic form of premature aging.[12] Mouse cells deficient for maturation of prelamin A show increased DNA damage and chromosome aberrations and are more sensitive to DNA damaging agents.[17] The inability to adequately repair DNA damages when A-type lamins are defective is likely responsible for some of the aspects of premature aging.
Diagnosis
Types of known laminopathies and other nuclear envelopathies
| Syndrome | OMIM ID | Symptoms | Mutation in | Identified in |
|---|---|---|---|---|
| Atypical Werner syndrome | 277700 | Progeria with increased severity compared to normal Werner syndrome | Lamin A/C | 2003[18] |
| Barraquer–Simons syndrome | 608709 | Lipodystrophy | Lamin B2 | 2006[8] |
| Buschke–Ollendorff syndrome | 166700 | Skeletal dysplasia, skin lesions | LEM domain containing protein 3 (lamin-binding protein) | 2004[19] |
| Cardiomyopathy, dilated, with quadriceps myopathy | 607920 | Cardiomyopathy | Lamin A/C | 2003[20] |
| Charcot–Marie–Tooth disease, axonal, type 2B1 | 605588 | Neuropathy | Lamin A/C | 2002[21] |
| Emery–Dreifuss muscular dystrophy, X-linked (EDMD) | 310300 | Skeletal and cardiac muscular dystrophy | Emerin (lamin-binding protein) | 1996,[22] 2000[23] |
| Emery–Dreifuss muscular dystrophy, autosomal dominant (EDMD2) | 181350 | Skeletal and cardiac muscular dystrophy | Lamin A/C | 1999[24] |
| Emery–Dreifuss muscular dystrophy, autosomal recessive (EDMD3) | 604929 | Skeletal and cardiac muscular dystrophy | Lamin A/C | 2000[25] |
| Familial partial lipodystrophy of the Dunnigan type (FPLD) | 151660 | Lipoatrophic diabetes | Lamin A/C | 2002[26] |
| Greenberg dysplasia | 215140 | Skeletal dysplasia | Lamin B receptor | 2003[27] |
| Hutchinson–Gilford progeria syndrome (HGPS) | 176670 | Progeria | Lamin A/C | 2003[12] |
| Leukodystrophy, demyelinating, adult-onset, autosomal dominant (ADLD) | 169500 | Progressive demyelinating disorder affecting the central nervous system | Lamin B1 (tandem gene duplication) | 2006[14] |
| Limb-girdle muscular dystrophy type 1B (LGMD1B) | 159001 | Muscular dystrophy of hips and shoulders, cardiomyopathy | Lamin A/C | 2000[28] |
| Lipoatrophy with diabetes, hepatic steatosis, hypertrophic cardiomyopathy, and leukomelanodermic papules (LDHCP) | 608056 | Lipoatrophic diabetes, fatty liver, hypertrophic cardiomyopathy, skin lesions | Lamin A/C | 2003[29] |
| Mandibuloacral dysplasia with type A lipodystrophy (MADA) | 248370 | Dysplasia and lipodystrophy | Lamin A/C | 2002[9] |
| Mandibuloacral dysplasia with type B lipodystrophy (MADB) | 608612 | Dysplasia and lipodystrophy | Zinc metalloprotease STE24 (prelamin-processing enzyme) | 2003[30] |
| Pelger–Huet anomaly (PHA) | 169400 | Myelodysplasia | Lamin B receptor | 2002[31] |
| Restrictive dermopathy, lethal | 275210 | Dermopathy | Lamin A/C or Zinc metalloprotease STE24 (prelamin-processing enzyme) | 2004 |
Treatment
Currently, there is no cure for laminopathies and treatment is largely symptomatic and supportive. Physical therapy and/or corrective orthopedic surgery may be helpful for patients with muscular dystrophies. Laminopathies affecting heart muscle may cause heart failure requiring treatment with medications including ACE inhibitors, beta blockers and aldosterone antagonists, while the abnormal heart rhythms that frequently occur in these patients may require a pacemaker or implantable defibrillator.[32] Treatment for neuropathies may include medication for seizures and spasticity.
Research
The recent progress in uncovering the molecular mechanisms of toxic progerin formation in laminopathies leading to premature aging has opened up the potential for the development of targeted treatment. The farnesylation of prelamin A and its pathological form progerin is carried out by the enzyme farnesyl transferase. Farnesyl transferase inhibitors (FTIs) can be used effectively to reduce symptoms in two mouse model systems for progeria and to revert the abnormal nuclear morphology in progeroid cell cultures. Two oral FTIs, lonafarnib and tipifarnib, are already in use as anti-tumor medication in humans and may become avenues of treatment for children suffering from laminopathic progeria. Nitrogen-containing bisphosphate drugs used in the treatment of osteoporosis reduce farnesyldiphosphate production and thus prelamin A farnesylation. Testing of these drugs may prove them to be useful in treating progeria as well. The use of antisense oligonucleotides to inhibit progerin synthesis in affected cells is another avenue of current research into the development of anti-progerin drugs.[33][34]
References
- Paradisi M, McClintock D, Boguslavsky RL, Pedicelli C, Worman HJ, Djabali K (2005). "Dermal fibroblasts in Hutchinson–Gilford progeria syndrome with the lamin A G608G mutation have dysmorphic nuclei and are hypersensitive to heat stress". BMC Cell Biol. 6: 27. doi:10.1186/1471-2121-6-27. PMC 1183198. PMID 15982412.
- Nagano A, Arahata K (2000). "Nuclear envelope proteins and associated diseases". Curr. Opin. Neurol. 13 (5): 533–9. doi:10.1097/00019052-200010000-00005. PMID 11073359.
- Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K (2004). "Lamin B1 is required for mouse development and nuclear integrity". Proc. Natl. Acad. Sci. U.S.A. 101 (28): 10428–33. Bibcode:2004PNAS..10110428V. doi:10.1073/pnas.0401424101. PMC 478588. PMID 15232008.
- Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, Durrington PN (2006). "Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy". Am J Hum Genet. 79: 383–389. doi:10.1086/505885. PMC 1559499. PMID 16826530.CS1 maint: multiple names: authors list (link)
- Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptácek LJ, Fu YH (2006). "Lamin B1 duplications cause autosomal dominant leukodystrophy". Nat Genet. 38: 1114–1123. doi:10.1038/ng1872. PMID 16951681.CS1 maint: multiple names: authors list (link)
- Mans BJ, Anantharaman V, Aravind L, Koonin EV (2004). "Comparative genomics, evolution and origins of the nuclear envelope and nuclear pore complex". Cell Cycle. 3 (12): 1612–37. doi:10.4161/cc.3.12.1316. PMID 15611647.
- Houben F, Ramaekers FC, Snoeckx LH, Broers JL (May 2007). "Role of nuclear lamina-cytoskeleton interactions in the maintenance of cellular strength". Biochim. Biophys. Acta. 1773 (5): 675–86. doi:10.1016/j.bbamcr.2006.09.018. PMID 17050008.
- Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, Durrington PN (2006). "Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy". Am. J. Hum. Genet. 79 (2): 383–9. doi:10.1086/505885. PMC 1559499. PMID 16826530.
- Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D'Apice MR, Massart C, Capon F, Sbraccia P, Federici M, Lauro R, Tudisco C, Pallotta R, Scarano G, Dallapiccola B, Merlini L, Bonne G (2002). "Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C". Am. J. Hum. Genet. 71 (2): 426–31. doi:10.1086/341908. PMC 379176. PMID 12075506.
- Zirn B, Kress W, Grimm T, Berthold LD, et al. (2008). "Association of homozygous LMNA mutation R471C with new phenotype: mandibuloacral dysplasia, progeria, and rigid spine muscular dystrophy". Am J Med Genet A. 146A (8): 1049–1054. doi:10.1002/ajmg.a.32259. PMID 18348272.
- Al-Haggar M, Madej-Pilarczyk A, Kozlowski L, Bujnicki JM, Yahia S, Abdel-Hadi D, Shams A, Ahmad N, Hamed S, Puzianowska-Kuznicka M (2012). "A novel homozygous p.Arg527Leu LMNA mutation in two unrelated Egyptian families causes overlapping mandibuloacral dysplasia and progeria syndrome". Eur J Hum Genet. 20 (11): 1134–40. doi:10.1038/ejhg.2012.77. PMC 3476705. PMID 22549407.
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS (2003). "Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome". Nature. 423 (6937): 293–8. Bibcode:2003Natur.423..293E. doi:10.1038/nature01629. hdl:2027.42/62684. PMID 12714972.
- Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH (2006). "Lamin B1 duplications cause autosomal dominant leukodystrophy". Nature Genetics. 38 (10): 1114–1123. doi:10.1038/ng1872. PMID 16951681.
- Lassoued K, Guilly MN, Danon F, Andre C, Dhumeaux D, Clauvel JP, Brouet JC, Seligmann M, Courvalin JC (1988). "Antinuclear autoantibodies specific for lamins. Characterization and clinical significance". Ann Intern Med. 108: 829–3. doi:10.7326/0003-4819-108-6-829.CS1 maint: multiple names: authors list (link)
- Redwood AB, Perkins SM, Vanderwaal RP, Feng Z, Biehl KJ, Gonzalez-Suarez I, Morgado-Palacin L, Shi W, Sage J, Roti-Roti JL, Stewart CL, Zhang J, Gonzalo S (2011). "A dual role for A-type lamins in DNA double-strand break repair". Cell Cycle. 10 (15): 2549–60. doi:10.4161/cc.10.15.16531. PMC 3180193. PMID 21701264.
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadiñanos J, López-Otín C, Tse HF, Hutchison C, Chen J, Cao Y, Cheah KS, Tryggvason K, Zhou Z (2005). "Genomic instability in laminopathy-based premature aging". Nat. Med. 11 (7): 780–5. doi:10.1038/nm1266. PMID 15980864.
- Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg E, Hanson NB, Martin GM, Mian IS, Kennedy BK, Oshima J (2003). "LMNA mutations in atypical Werner's syndrome". Lancet. 362 (9382): 440–5. doi:10.1016/S0140-6736(03)14069-X. PMID 12927431.
- Hellemans J, Preobrazhenska O, Willaert A, Debeer P, Verdonk PCM, Costa T, Janssens K, Menten B, Van Roy N, Vermeulen SJT, Savarirayan R, Van Hul W, et al. (2004). "Loss-of-function mutations in LEMD3 result in osteopoikilosis, Buschke–Ollendorff syndrome and melorheostosis". Nature Genetics. 36 (11): 1213–8. doi:10.1038/ng1453. PMID 15489854.
- Charniot JC, Pascal C, Bouchier C, Sebillon P, Salama J, Duboscq-Bidot L, Peuchmaurd M, Desnos M, Artigou JY, Komajda M (2003). "Functional consequences of an LMNA mutation associated with a new cardiac and non-cardiac phenotype". Hum. Mutat. 21 (5): 473–81. doi:10.1002/humu.10170. PMID 12673789.
- De Sandre-Giovannoli A, Chaouch M, Kozlov S, Vallat JM, Tazir M, Kassouri N, Szepetowski P, Hammadouche T, Vandenberghe A, Stewart CL, Grid D, Levy N (2002). "Homozygous defects in LMNA, encoding lamin A/C nuclear-envelope proteins, cause autosomal recessive axonal neuropathy in human (Charcot–Marie–Tooth disorder type 2) and mouse". Am. J. Hum. Genet. 70 (3): 726–36. doi:10.1086/339274. PMC 384949. PMID 11799477.
- Manilal S, Nguyen TM, Sewry CA, Morris GE (1996). "The Emery–Dreifuss muscular dystrophy protein, emerin, is a nuclear membrane protein". Hum. Mol. Genet. 5 (6): 801–8. doi:10.1093/hmg/5.6.801. PMID 8776595.
- Clements L, Manilal S, Love DR, Morris GE (2000). "Direct interaction between emerin and lamin A". Biochem. Biophys. Res. Commun. 267 (3): 709–14. doi:10.1006/bbrc.1999.2023. PMID 10673356.
- Bonne G, Di Barletta MR, Varnous S, Becane HM, Hammouda EH, Merlini L, Muntoni F, Greenberg CR, Gary F, Urtizberea JA, Duboc D, Fardeau M, Toniolo D, Schwartz K (1999). "Mutations in the gene encoding lamin A/C cause autosomal dominant Emery–Dreifuss muscular dystrophy". Nature Genetics. 21 (3): 285–8. doi:10.1038/6799. PMID 10080180.
- Raffaele di Barletta M, Ricci E, Galluzzi G, Tonali P, Mora M, Morandi L, Romorini A, Voit T, Orstavik KH, Merlini L, Trevisan C, Biancalana V, Housmanowa-Petrusewicz I, Bione S, Ricotti R, Schwartz K, Bonne G, Toniolo D (2000). "Different mutations in the LMNA gene cause autosomal dominant and autosomal recessive Emery–Dreifuss muscular dystrophy". Am. J. Hum. Genet. 66 (4): 1407–12. doi:10.1086/302869. PMC 1288205. PMID 10739764.
- Cao H, Hegele RA (2002). "Nuclear lamin A/C R482Q mutation in Canadian kindreds with Dunnigan-type familial partial lipodystrophy". Hum. Mol. Genet. 9 (1): 109–12. doi:10.1093/hmg/9.1.109. PMID 10587585.
- Waterham HR, Koster J, Mooyer P, van Noort G, Kelley RI, Wilcox WR, Wanders RJ, Hennekam RC, Oosterwijk JC (2003). "Autosomal recessive HEM/Greenberg skeletal dysplasia is caused by 3-beta-hydroxysterol delta(14)-reductase deficiency due to mutations in the lamin B receptor gene". Am. J. Hum. Genet. 72 (4): 1013–17. doi:10.1086/373938. PMC 1180330. PMID 12618959.
- Muchir A, Bonne G, van der Kooi AJ, van Meegen M, Baas F, Bolhuis PA, de Visser M, Schwartz K (2000). "Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B)". Hum. Mol. Genet. 9 (9): 1453–9. doi:10.1093/hmg/9.9.1453. PMID 10814726.
- Caux F, Dubosclard E, Lascols O, Buendia B, Chazouilleres O, Cohen A, Courvalin JC, Laroche L, Capeau J, Vigouroux C, Christin-Maitre S (2003). "A new clinical condition linked to a novel mutation in lamins A and C with generalized lipoatrophy, insulin-resistant diabetes, disseminated leukomelanodermic papules, liver steatosis, and cardiomyopathy". J. Clin. Endocrinol. Metab. 88 (3): 1006–13. doi:10.1210/jc.2002-021506. PMID 12629077.
- Agarwal AK, Fryns JP, Auchus RJ, Garg A (2003). "Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia". Hum. Mol. Genet. 12 (16): 1995–2001. doi:10.1093/hmg/ddg213. PMID 12913070.
- Hoffmann K, Dreger CK, Olins AL, Olins DE, Shultz LD, Lucke B, Karl H, Kaps R, Muller D, Vaya A, Aznar J, Ware RE, Cruz NS, Lindner TH, Herrmann H, Reis A, Sperling K (2002). "Mutations in the gene encoding the lamin B receptor produce an altered nuclear morphology in granulocytes (Pelger-Huet anomaly)". Nature Genetics. 31 (4): 410–4. doi:10.1038/ng925. PMID 12118250.
- Captur, Gabriella; Arbustini, Eloisa; Bonne, Gisèle; Syrris, Petros; Mills, Kevin; Wahbi, Karim; Mohiddin, Saidi A.; McKenna, William J.; Pettit, Stephen (2017-11-25). "Lamin and the heart". Heart. 104 (6): 468–479. doi:10.1136/heartjnl-2017-312338. ISSN 1468-201X. PMID 29175975.
- Rusinal AE, Sinensky MS (2006). "Farnesylated lamins, progeroid syndromes and farnesyl transferase inhibitors". J. Cell Sci. 119 (Pt 16): 3265–72. doi:10.1242/jcs.03156. PMID 16899817.
- Meta M, Yang SH, Bergo MO, Fong LG, Young SG (2006). "Protein farnesyltransferase inhibitors and progeria". Trends Mol. Med. 12 (10): 480–7. doi:10.1016/j.molmed.2006.08.006. PMID 16942914.