Frontotemporal dementia
The frontotemporal dementias (FTD) encompass six types of dementia involving the frontal or temporal lobes. They are: behavioral variant of FTD, semantic variant primary progressive aphasia, nonfluent agrammatic variant primary progressive aphasia, corticobasal syndrome, progressive supranuclear palsy, and FTD associated with motor neuron disease.[1]
| Frontotemporal dementia | |
|---|---|
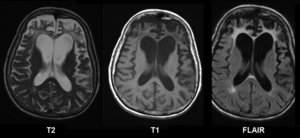 | |
| Brain MRI of a female 65 y.o. white patient with Pick's disease. Cortex and white matter atrophy of the frontal lobes is clearly visible in all images. | |
| Specialty | Psychiatry, neurology |
| Causes | frontotemporal lobar degeneration |
One variant is the clinical presentation of frontotemporal lobar degeneration, which is characterized by progressive neuronal loss predominantly involving the frontal or temporal lobes, and typical loss of over 70% of spindle neurons, while other neuron types remain intact.[2]
It was first described by Arnold Pick in 1892 and was originally called "Pick's disease", a term now reserved for Pick disease, one specific type of frontotemporal dementia.[3] Second only to Alzheimer's disease (AD) in prevalence, FTD accounts for 20% of young-onset dementia cases.[4] Signs and symptoms typically manifest in late adulthood, more commonly between the ages of 45 and 65, approximately equally affecting men and women.[4]
Common signs and symptoms include significant changes in social and personal behavior, apathy, blunting of emotions, and deficits in both expressive and receptive language. Currently, there is no cure for FTD, but there are treatments that help alleviate symptoms.
Signs and symptoms
Frontotemporal dementia (FTD) classically affects adults in their fifth to sixth decade of life. These patients usually describe a gradual onset and progression of changes in behavior or language deficits for several years prior to presentation to a neurologist.[1]
FTD is traditionally difficult to diagnose due to the heterogeneity of the associated symptoms. Signs and symptoms are classified into three groups based on the functions of the frontal and temporal lobes:[3]
- Behavioural variant frontotemporal dementia (BvFTD) is characterized by changes in social behavior and conduct, with loss of social awareness and poor impulse control.[5]
- Semantic dementia (SD) is characterized by the loss of semantic understanding, resulting in impaired word comprehension, although speech remains fluent and grammatically faultless.[5]
- Progressive nonfluent aphasia (PNFA) is characterized by progressive difficulties in speech production.[5]
However, the following abilities in the person with FTD are preserved:[4]
- Perception
- Spatial skills
- Memory
- Praxis (motor planning to perform tasks or movements)
In later stages of FTD, the clinical phenotypes may overlap.[5] FTD patients tend to struggle with binge eating and compulsive behaviors.[6] These binge eating habits are often associated with abnormal eating behavior including overeating, stuffing oneself with food, changes in food preferences (cravings for more sweets, carbohydrates), eating inedible objects and snatching food from others. Recent findings from structural MRI research have indicated that eating changes in FTD are associated with atrophy (wasting) in the right ventral insula, striatum, and orbitofrontal cortex.[6]
Patients with FTD show marked deficiencies in executive functioning and working memory.[7] Most FTD patients become unable to perform skills that require complex planning or sequencing.[8] In addition to the characteristic cognitive dysfunction, a number of primitive reflexes known as frontal release signs are often able to be elicited. Usually the first of these frontal release signs to appear is the palmomental reflex which appears relatively early in the disease course whereas the palmar grasp reflex and rooting reflex appear late in the disease course.
In rare cases, FTD can occur in patients with motor neuron disease (MND) (typically amyotrophic lateral sclerosis). The prognosis for people with MND is worse when combined with FTD, shortening survival by about a year.[9]
Genetics
A higher proportion of FTD cases seem to have a familial component than more common neurodegenerative diseases like Alzheimer's disease. More and more mutations and genetic variants are being identified all the time, so the lists of genetic influences require consistent updating.
- Tau-positive frontotemporal dementia with parkinsonism (FTDP-17) is caused by mutations in the MAPT gene on chromosome 17 that encodes the Tau protein[10] It has been determined that there is a direct relationship between the type of tau mutation and the neuropathology of gene mutations. The mutations at the splice junction of exon 10 of tau lead to the selective deposition of the repetitive tau in neurons and glia. The pathological phenotype associated with mutations elsewhere in tau is less predictable with both typical neurofibrillary tangles (consisting of both 3 repeat and 4 repeat tau) and Pick bodies (consisting of 3 repeat tau) having been described. The presence of tau deposits within glia is also variable in families with mutations outside of exon 10. This disease is now informally designated FTDP-17T. FTD shows a linkage to the region of the tau locus on chromosome 17, but it is believed that there are two loci leading to FTD within megabases of each other on chromosome 17.[11]
- FTD caused by FTLD-TDP43 has numerous genetic causes. Some cases are due to mutations in the GRN gene, also located on chromosome 17. Others are caused by VCP mutations, although these patients present with a complex picture of multisystem proteinopathy that can include amyotrophic lateral sclerosis, inclusion body myopathy, Paget's disease of bone, and FTD. The most recent addition to the list is a hexanucleotide repeat expansion in intron 1 of C9ORF72.[12] Only one or two cases have been reported describing TARDBP (the TDP-43 gene) mutations in a clinically pure FTD (FTD without MND).
- Several other genes have been linked to this condition. These include CYLD, OPTN, SQSTM1 and TBK1.[13] These genes have been implicated in the autophagy pathway.
- No genetic causes of FUS pathology in FTD have yet been reported.
Pathology
There are three main histological subtypes found at post-mortem: FTLD-tau, FTLD-TDP, and FTLD-FUS. Dementia lacking distinctive histology (DLDH) is a rare and controversial entity. New analysis has allowed many cases previously described as DLDH to be reclassified into one of the positively defined subgroups. In rare cases, patients with clinical FTD were found to have changes consistent with Alzheimer's disease on autopsy.[14] The most severe brain atrophy appears to be associated with Pick's disease, corticobasal degeneration, and TDP pathology associated with behavioral-variant FTD.[15]
With regard to the genetic defects that have been found, repeat expansion in the C9orf72 gene is considered a major contribution to frontotemporal lobar degeneration, although defects in the GRN and MAPT genes are also associated with it.[16]
Diagnosis
Structural MRI scans often reveal frontal lobe and/or anterior temporal lobe atrophy but in early cases the scan may seem normal. Atrophy can be either bilateral or asymmetric.[4] Registration of images at different points of time (e.g., one year apart) can show evidence of atrophy that otherwise (at individual time points) may be reported as normal. Many research groups have begun using techniques such as magnetic resonance spectroscopy, functional imaging and cortical thickness measurements in an attempt to offer an earlier diagnosis to the FTD patient. Fluorine-18-fluorodeoxyglucose positron emission tomography (FDG-PET) scans classically show frontal and/or anterior temporal hypometabolism, which helps differentiate the disease from Alzheimer's disease. The PET scan in Alzheimer's disease classically shows biparietal hypometabolism. Meta-analyses based on imaging methods have shown that frontotemporal dementia mainly affects a frontomedial network discussed in the context of social cognition or 'theory of mind'.[17] This is entirely in keeping with the notion that on the basis of cognitive neuropsychological evidence, the ventromedial prefrontal cortex is a major locus of dysfunction early on in the course of the behavioural variant of frontotemporal degeneration.[18] The language subtypes of frontotemporal lobar degeneration (semantic dementia and progressive nonfluent aphasia) can be regionally dissociated by imaging approaches in vivo.[19]
The confusion between Alzheimer's and FTD is justifiable due to the similarities between their initial symptoms. Patients do not have difficulty with movement and other motor tasks.[20] As FTD symptoms appear, it is difficult to differentiate between a diagnosis of Alzheimer's disease and FTD. There are distinct differences in the behavioral and emotional symptoms of the two dementias, notably, the blunting of emotions seen in FTD patients.[4] In the early stages of FTD, anxiety and depression are common, which may result in an ambiguous diagnosis. However, over time, these ambiguities fade away as this dementia progresses and defining symptoms of apathy, unique to FTD, start to appear.
Recent studies over several years have developed new criteria for the diagnosis of behavioral variant frontotemporal dementia (bvFTD). Six distinct clinical features have been identified as symptoms of bvFTD.[21]
- Disinhibition
- Apathy/Inertia
- Loss of Sympathy/Empathy
- Perseverative/compulsive behaviors
- Hyperorality
- Dysexecutive neuropsychological profile
Of the six features, three must be present in a patient to diagnose one with possible bvFTD. Similar to standard FTD, the primary diagnosis stems from clinical trials that identify the associated symptoms, instead of imaging studies.[21] The above criteria are used to distinguish bvFTD from disorders such as Alzheimer's and other causes of dementia. In addition, the new criteria allow for a diagnostic hierarchy distinguished possible, probable, and definite bvFTD based on the number of symptoms present.
Neuropsychological tests
The progression of the degeneration caused by bvFTD may follow a predictable course. The degeneration begins in the orbitofrontal cortex and medial aspects such as ventromedial cortex. In later stages, it gradually expands its area to the dorsolateral cortex and the temporal lobe.[22] Thus, the detection of dysfunction of the orbitofrontal cortex and ventromedial cortex is important in the detection of early stage bvFTD. As stated above, a behavioural change may occur before the appearance of any atrophy in the brain in the course of the disease. Because of that, image scanning such as MRI can be insensitive to the early degeneration and it is difficult to detect early-stage bvFTD.
In neuropsychology, there is an increasing interest in using neuropsychological tests such as the Iowa gambling task or Faux Pas Recognition test as an alternative to imaging for the diagnosis of bvFTD.[23] Both the Iowa gambling task and the Faux Pas test are known to be sensitive to dysfunction of the orbitofrontal cortex.
Faux Pas Recognition test is intended to measure one’s ability to detect faux pas types of social blunders (accidentally make a statement or an action that offends others). It is suggested that people with orbitofrontal cortex dysfunction show a tendency to make social blunders due to a deficit in self-monitoring.[24] Self-monitoring is the ability of individuals to evaluate their behaviour to make sure that their behaviour is appropriate in particular situations. The impairment in self-monitoring leads to a lack of social emotion signals. The social emotions such as embarrassment are important in the way that they signal the individual to adapt social behaviour in an appropriate manner to maintain relationships with others. Though patients with damage to the OFC retain intact knowledge of social norms, they fail to apply it to actual behaviour because they fail to generate social emotions that promote adaptive social behaviour.[24]
The other test, the Iowa gambling task, is a psychological test intended to simulate real-life decision making. The underlying concept of this test is the somatic marker hypothesis. This hypothesis argues that when people have to make complex uncertain decisions, they employ both cognitive and emotional processes to assess the values of the choices available to them. Each time a person makes a decision, both physiological signals and evoked emotion (somatic marker) are associated with their outcomes and it accumulates as experience. People tend to choose the choice which might produce the outcome reinforced with positive stimuli, thus it biases decision-making towards certain behaviours while avoiding others.[25] It is thought that somatic marker is processed in orbitofrontal cortex.
The symptoms observed in bvFTD are caused by dysfunction of the orbitofrontal cortex, thus these two neuropsychological tests might be useful in detecting the early stage bvFTD. However, as self-monitoring and somatic marker processes are so complex, it likely involves other brain regions. Therefore, neuropsychological tests are sensitive to the dysfunction of orbitofrontal cortex, yet not specific to it. The weakness of these tests is that they do not necessarily show dysfunction of the orbitofrontal cortex.
In order to solve this problem, some researchers combined neuropsychological tests which detect the dysfunction of orbitofrontal cortex into one so that it increases its specificity to the degeneration of the frontal lobe in order to detect the early-stage bvFTD. They invented the Executive and Social Cognition Battery which comprises five neuropsychological tests.
- Iowa gambling task
- Faux Pas test
- Hotel task
- Mind in the Eyes
- Multiple Errands Task
The result has shown that this combined test is more sensitive in detecting the deficits in early bvFTD.[23]
Management
Currently, there is no cure for FTD. Treatments are available to manage the behavioral symptoms. Disinhibition and compulsive behaviors can be controlled by selective serotonin reuptake inhibitors (SSRIs).[26][27] Although Alzheimer's and FTD share certain symptoms, they cannot be treated with the same pharmacological agents because the cholinergic systems are not affected in FTD.[4]
Because FTD often occurs in younger people (i.e. in their 40s or 50s), it can severely affect families. Patients often still have children living in the home. Financially, it can be devastating as the disease strikes at the time of life that often includes the top wage-earning years.
Prognosis
Symptoms of frontotemporal dementia progress at a rapid, steady rate. Patients suffering from the disease can survive for 2–20 years. Eventually patients will need 24-hour care for daily function.[28]
CSF leaks are a known cause of reversible frontotemporal dementia.[29]
History
Frontotemporal dementia was first described by Pick in 1892.[30] In 1989, Snowden suggested the term “semantic dementia” to describe the patient with predominant left temporal atrophy and aphasia that Pick described. The first research criteria for FTD “Clinical and neuropathological criteria for frontotemporal dementia. The Lund and Manchester Groups,” was developed in 1994. The clinical diagnostic criteria were revised in the late 1990s, when the FTD spectrum was divided into a behavioral variant, a nonfluent aphasia variant and a semantic dementia variant.[31] The most recent revision of the clinical research criteria was by International Behavioural Variant FTD Criteria Consortium (FTDC) in 2011.[32]
See also
- Alcoholic dementia
- Alzheimer disease
- Aphasia
- Lewy body dementia
- Logopenic progressive aphasia
- Primary progressive aphasia
- Proteopathy
- Vascular dementia
References
- Finger, EC (April 2016). "Frontotemporal Dementias". Continuum (Minneapolis, Minn.). 22 (2 Dementia): 464–89. doi:10.1212/CON.0000000000000300. PMC 5390934. PMID 27042904.
- "Brain Cells for Socializing". Smithsonian. Retrieved 30 October 2015.
- Cardarelli R, Kertesz A, Knebl JA (December 2010). "Frontotemporal dementia: a review for primary care physicians". Am Fam Physician. 82 (11): 1372–7. PMID 21121521.
- Snowden JS, Neary D, Mann DM (February 2002). "Frontotemporal dementia". Br J Psychiatry. 180 (2): 140–3. doi:10.1192/bjp.180.2.140. PMID 11823324.
- Sleegers, Kristel; Cruts, Marc; Van Broeckhoven, Christine (2010). "Molecular Pathways of Frontotemporal Lobar Degeneration". Annual Review of Neuroscience. 33 (1): 71–88. doi:10.1146/annurev-neuro-060909-153144. PMID 20415586.
- Piguet O (November 2011). "Eating disturbance in behavioural-variant frontotemporal dementia". J. Mol. Neurosci. 45 (3): 589–93. doi:10.1007/s12031-011-9547-x. PMID 21584651.
- Neary D, Snowden J, Mann D (2005). "Frontotemporal dementia". Lancet Neurol. 4 (11): 771–80. doi:10.1016/s1474-4422(05)70223-4. PMID 16239184.
- Kramer JH, Jurik J, Sha SJ, et al. (December 2003). "Distinctive neuropsychological patterns in frontotemporal dementia, semantic dementia, and Alzheimer disease". Cogn Behav Neurol. 16 (4): 211–8. doi:10.1097/00146965-200312000-00002. PMID 14665820.
- Olney RK, Murphy J, Forshew D, et al. (December 2005). "The effects of executive and behavioral dysfunction on the course of ALS". Neurology. 65 (11): 1774–7. doi:10.1212/01.wnl.0000188759.87240.8b. PMID 16344521.
- Luc Buée; André Delacourte (1999). "Comparative Biochemistry of Tau in Progressive Supranuclear Palsy, Corticobasal Degeneration, FTDP-17 and Pick's Disease". Brain Pathology. 9 (4): 681–693. doi:10.1111/j.1750-3639.1999.tb00550.x. PMID 10517507.
- Hardy, John; Momeni, Parastoo; Traynor, Bryan J. (April 2006). "Frontal temporal dementia: dissecting the aetiology and pathogenesis" (PDF). Brain. 26. 129 (4): 830–831. doi:10.1093/brain/awl035. PMID 16543401. Retrieved July 8, 2017.
- Convery R, Mead S, Rohrer JD (2018) Clinical, genetic and neuroimaging features of frontotemporal dementia. Neuropathol Appl Neurobiol
- Dobson-Stone C, Hallupp M, Shahheydari H, Ragagnin AMG, Chatterton Z, Carew-Jones F, Shepherd CE, Stefen H, Paric E, Fath T, Thompson EM, Blumbergs P, Short CL, Field CD, Panegyres PK, Hecker J, Nicholson G, Shaw AD, Fullerton JM, Luty AA, Schofield PR, Brooks WS, Rajan N, Bennett MF, Bahlo M, Landers JE, Piguet O, Hodges JR, Halliday GM, Topp SD, Smith BN, Shaw CE, McCann E, Fifita JA, Williams KL, Atkin JD, Blair IP, Kwok JB (2020). "CYLD is a causative gene for frontotemporal dementia - amyotrophic lateral sclerosis". Brain. 143 (3): 783–799. doi:10.1093/brain/awaa039. PMID 32185393.CS1 maint: multiple names: authors list (link)
- Liscic RM, Storandt M, Cairns NJ, Morris JC (April 2007). "Clinical and psychometric distinction of frontotemporal and Alzheimer dementias". Arch. Neurol. 64 (4): 535–40. doi:10.1001/archneur.64.4.535. PMID 17420315.
- Jonathan D. Rohrer; Tammaryn Lashley; Jonathan M. Schott; Jane E. Warren; Simon Mead; Adrian M. Isaacs; Jonathan Beck; John Hardy; Rohan de Silva; Elizabeth Warrington; Claire Troakes; Safa Al-Sarraj; Andrew King; Barbara Borroni; Matthew J. Clarkson; Sebastien Ourselin; Janice L. Holton; Nick C. Fox; Tamas Revesz; Martin N. Rossor & Jason D. Warren (corresponding author) (September 2011). "Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration". Brain. 134 (9): 2565–2581. doi:10.1093/brain/awr198. PMC 3170537. PMID 21908872.
- van der Zee, Julie; Van Broeckhoven, Christine (7 January 2014). "Dementia in 2013: Frontotemporal lobar degeneration—building on breakthroughs". Nature Reviews Neurology. 10 (2): 70–72. doi:10.1038/nrneurol.2013.270. PMID 24394289.
- Schroeter ML, Raczka KK, Neumann J, von Cramon DY (2008). "Neural networks in frontotemporal dementia – A meta-analysis". Neurobiology of Aging. 29 (3): 418–426. doi:10.1016/j.neurobiolaging.2006.10.023. PMID 17140704.
- Rahman S, Sahakian BJ, Hodges JR, Rogers RD, Robbins TW (August 1999). "Specific cognitive deficits in mild frontal variant frontotemporal dementia" (PDF). Brain. 122 (Pt 8): 1469–93. doi:10.1093/brain/122.8.1469. PMID 10430832.
- Schroeter ML, Raczka KK, Neumann J, von Cramon DY (2007). "Towards a nosology for frontotemporal lobar degenerations – A meta-analysis involving 267 subjects". NeuroImage. 36 (3): 497–510. doi:10.1016/j.neuroimage.2007.03.024. PMID 17478101.
- Steinbart EJ, Smith CO, Poorkaj P, Bird TD (November 2001). "Impact of DNA testing for early-onset familial Alzheimer disease and frontotemporal dementia". Arch. Neurol. 58 (11): 1828–31. doi:10.1001/archneur.58.11.1828. PMID 11708991.
- Rascovsky K, Hodges JR, Knopman D, et al. (September 2011). "Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia" (PDF). Brain. 134 (Pt 9): 2456–77. doi:10.1093/brain/awr179. PMC 3170532. PMID 21810890.
- Krueger, C.E.; Bird, A.C.; Growdon, M.E.; Jang, J.Y.; Miller, B.L. & Kramer, J.K. (2009). "Conflict in monitoring early frontotemporal dementia". Neurology. 73 (5): 349–55. doi:10.1212/wnl.0b013e3181b04b24. PMC 2725928. PMID 19652138.
- Torralva, T; Roca, M; Gleichgerrcht, E; Bekinschtein, T & Manes, F (2009). "A neuropsychological battery to detect specific executive and social cognitive impairments in early frontotemporal dementia" (PDF). Brain. 132 (5): 1299–1309. doi:10.1093/brain/awp041. PMID 19336463.
- Beer, J.S.; John, O.P.; Scabini, D. & Knight, R.T. (2006). "Orbitofrontal cortex and Social behaviour: Integrating Self-monitoring and Emotion-cognition interactions". Journal of Cognitive Neuroscience. 18 (6): 871–888. CiteSeerX 10.1.1.527.3607. doi:10.1162/jocn.2006.18.6.871. PMID 16839295.
- Damasio, A.R.; Everitt, B. J.; Bishop, D. (29 October 1996). "The Somatic marker hypothesis and the possible functions of the prefrontal cortex". Phil. Trans. R. Soc. Lond. B. 351 (1346): 1413–20. doi:10.1098/rstb.1996.0125. PMID 8941953. S2CID 1841280.
- Swartz JR, Miller BL, Lesser IM, Darby AL (May 1997). "Frontotemporal dementia: treatment response to serotonin selective reuptake inhibitors". J Clin Psychiatry. 58 (5): 212–6. doi:10.4088/jcp.v58n0506. PMID 9184615.
- "Medications for behavioral symptoms". ucsf.edu. Retrieved 30 October 2015.
- Kertesz A (June 2004). "Frontotemporal dementia/Pick's disease". Arch. Neurol. 61 (6): 969–71. doi:10.1001/archneur.61.6.969. PMID 15210543.
- Samson, K. (2002). "Hypotension May Cause Frontotemporal Dementia". Neurology Today. 2 (9): 35–36. doi:10.1097/00132985-200209000-00013.
- PICK, A. (1892). "Uber die Beziehungen der senilen Hirnatrophie zur Aphasie". Prag Med Wochenschr. 17: 165–167.
- Olney, Nicholas T.; Spina, Salvatore; Miller, Bruce L. (May 2017). "Frontotemporal Dementia". Neurologic Clinics. 35 (2): 339–374. doi:10.1016/j.ncl.2017.01.008. ISSN 0733-8619. PMC 5472209. PMID 28410663.
- Rascovsky, Katya; Hodges, John R.; Knopman, David; Mendez, Mario F.; Kramer, Joel H.; Neuhaus, John; van Swieten, John C.; Seelaar, Harro; Dopper, Elise G. P.; Onyike, Chiadi U.; Hillis, Argye E. (September 2011). "Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia". Brain. 134 (9): 2456–2477. doi:10.1093/brain/awr179. ISSN 0006-8950. PMC 3170532. PMID 21810890.
Further reading
- Liu, W; Miller, B. L.; Kramer, J. H.; Rankin, K.; Wyss-Coray, C.; Gearhart, R.; Phengrasamy, L.; Weiner, M.; Rosen, H. J. (1 March 2004). "Behavioral disorders in the frontal and temporal variants of frontotemporal dementia". Neurology. 5. 62 (5): 742–748. doi:10.1212/01.WNL.0000113729.77161.C9. PMC 2367136. PMID 15007124.
- Hodges, J.R (2 April 2003). "A study of stereotypic behaviours in Alzheimer's disease and frontal and temporal variant frontotemporal dementia". Neurol Neurosurg Psychiatry. 74 (10): 1398–1402. doi:10.1136/jnnp.74.10.1398. PMC 1757381. PMID 14570833.
- Pagon RA, et al. (1993). "GRN-Related Frontotemporal Dementia". GeneReviews. PMID 20301545.
- Pagon RA, et al. (1993). "MAPT-Related Disorders including Frontotemporal Dementia with Parkinsonism-17 (FTDP-17)". GeneReviews. PMID 20301678.
|chapter=ignored (help) - Reynolds, Marilyn. 'Til Death or Dementia Do Us Part. River Rock Books, 2017.
External links
| Classification | |
|---|---|
| External resources |