Wound healing
Wound healing refers to a living organism's replacement of destroyed or damaged tissue by newly produced tissue.[1]
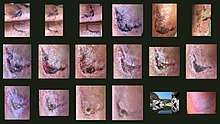
| Hand abrasion | ||||
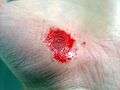 |
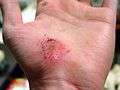 |
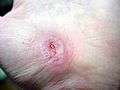 |
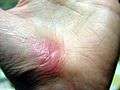 | |
| Approximate days since injury | ||||
| 0 | 3 | 17 | 30 | |
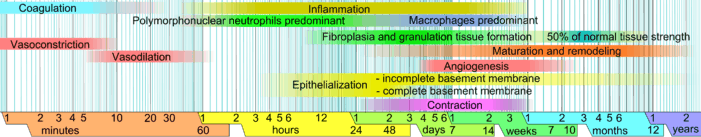
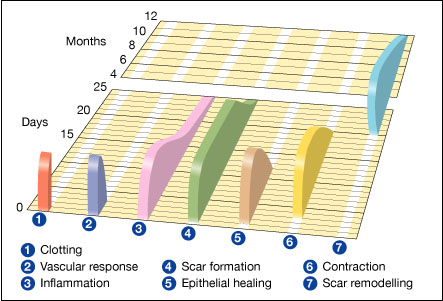
In this article, which focuses on humans, wound healing is depicted in a discrete timeline of physical attributes (phases) constituting the post-trauma repairing process. In undamaged skin, the epidermis (surface layer) and dermis (deeper layer) form a protective barrier against the external environment. When the barrier is broken, a regulated sequence of biochemical events is set into motion to repair the damage.[1][2] This process is divided into predictable phases: blood clotting (hemostasis), inflammation, tissue growth (cell proliferation), and tissue remodeling (maturation and cell differentiation). Blood clotting may be considered to be part of the inflammation stage instead of a separate stage.[3]
The wound healing process is not only complex but also fragile, and it is susceptible to interruption or failure leading to the formation of non-healing chronic wounds. Factors that contribute to non-healing chronic wounds are diabetes, venous or arterial disease, infection, and metabolic deficiencies of old age.[4]
Wound care encourages and speeds wound healing via cleaning and protection from reinjury or infection. Depending on each patient's needs, it can range from the simplest first aid to entire nursing specialties such as wound, ostomy, and continence nursing and burn center care.
Stages
- Hemostasis (blood clotting): Within the first few minutes of injury, platelets in the blood begin to stick to the injured site. This activates the platelets, causing a few things to happen. They change into an amorphous shape, more suitable for clotting, and they release chemical signals to promote clotting. This results in the activation of fibrin, which forms a mesh and acts as "glue" to bind platelets to each other. This makes a clot that serves to plug the break in the blood vessel, slowing/preventing further bleeding.[5][6]
- Inflammation: During this phase, damaged and dead cells are cleared out, along with bacteria and other pathogens or debris. This happens through the process of phagocytosis, where white blood cells "eat" debris by engulfing it. Platelet-derived growth factors are released into the wound that cause the migration and division of cells during the proliferative phase.
- Proliferation (growth of new tissue): In this phase, angiogenesis, collagen deposition, granulation tissue formation, epithelialization, and wound contraction occur.[7] In angiogenesis, vascular endothelial cells form new blood vessels.[8] In fibroplasia and granulation tissue formation, fibroblasts grow and form a new, provisional extracellular matrix (ECM) by excreting collagen and fibronectin.[7] Concurrently, re-epithelialization of the epidermis occurs, in which epithelial cells proliferate and 'crawl' atop the wound bed, providing cover for the new tissue.[9] In wound contraction, myofibroblasts decrease the size of the wound by gripping the wound edges and contracting using a mechanism that resembles that in smooth muscle cells. When the cells' roles are close to complete, unneeded cells undergo apoptosis.[7]
- Maturation (remodeling): During maturation and remodeling, collagen is realigned along tension lines, and cells that are no longer needed are removed by programmed cell death, or apoptosis.
Timing and re-epithelialization
Timing is important to wound healing. Critically, the timing of wound re-epithelialization can decide the outcome of the healing.[11] If the epithelization of tissue over a denuded area is slow, a scar will form over many weeks, or months;[12][13] If the epithelization of a wounded area is fast, the healing will result in regeneration.[13]
Early vs cellular phase
Wound healing is classically divided into hemostasis, inflammation, proliferation, and remodeling. Although a useful construct, this model employs considerable overlapping among individual phases. A complementary model has recently been described[1] where the many elements of wound healing are more clearly delineated. The importance of this new model becomes more apparent through its utility in the fields of regenerative medicine and tissue engineering (see Research and development section below). In this construct, the process of wound healing is divided into two major phases: the early phase and the cellular phase:[1]
The early phase, which begins immediately following skin injury, involves cascading molecular and cellular events leading to hemostasis and formation of an early, makeshift extracellular matrix that provides structural staging for cellular attachment and subsequent cellular proliferation.
The cellular phase involves several types of cells working together to mount an inflammatory response, synthesize granulation tissue, and restore the epithelial layer.[1] Subdivisions of the cellular phase are:
- Macrophages and inflammatory components (within 1–2 days)
- Epithelial-mesenchymal interaction: re-epithelialization (phenotype change within hours, migration begins on day 1 or 2)
- Fibroblasts and myofibroblasts: progressive alignment, collagen production, and matrix contraction (between day 4 and day 14)
- Endothelial cells and angiogenesis (begins on day 4)
- Dermal matrix: elements of fabrication (begins on day 4, lasting 2 weeks) and alteration/remodeling (begins after week 2, lasting weeks to months—depending on wound size).[1]
Inflammatory phase
Just before the inflammatory phase is initiated, the clotting cascade occurs in order to achieve hemostasis, or stop blood loss by way of a fibrin clot. Thereafter, various soluble factors (including chemokines and cytokines) are released to attract cells that phagocytise debris, bacteria, and damaged tissue, in addition to releasing signaling molecules that initiate the proliferative phase of wound healing.
Clotting cascade
When tissue is first wounded, blood comes in contact with collagen, triggering blood platelets to begin secreting inflammatory factors.[15] Platelets also express sticky glycoproteins on their cell membranes that allow them to aggregate, forming a mass.[7]
Fibrin and fibronectin cross-link together and form a plug that traps proteins and particles and prevents further blood loss.[16] This fibrin-fibronectin plug is also the main structural support for the wound until collagen is deposited.[7] Migratory cells use this plug as a matrix to crawl across, and platelets adhere to it and secrete factors.[7] The clot is eventually lysed and replaced with granulation tissue and then later with collagen.
Platelets, the cells present in the highest numbers shortly after a wound occurs, release mediators into the blood, including cytokines and growth factors.[15] Growth factors stimulate cells to speed their rate of division. Platelets release other proinflammatory factors like serotonin, bradykinin, prostaglandins, prostacyclins, thromboxane, and histamine,[3] which serve several purposes, including increasing cell proliferation and migration to the area and causing blood vessels to become dilated and porous. In many ways, extravasated platelets in trauma perform a similar function to tissue macrophages and mast cells exposed to microbial molecular signatures in infection: they become activated, and secrete molecular mediators – vasoactive amines, eicosanoids, and cytokines – that initiate the inflammatory process.
Vasoconstriction and vasodilation
Immediately after a blood vessel is breached, ruptured cell membranes release inflammatory factors like thromboxanes and prostaglandins that cause the vessel to spasm to prevent blood loss and to collect inflammatory cells and factors in the area.[3] This vasoconstriction lasts five to ten minutes and is followed by vasodilation, a widening of blood vessels, which peaks at about 20 minutes post-wounding.[3] Vasodilation is the end result of factors released by platelets and other cells. The main factor involved in causing vasodilation is histamine.[3][15] Histamine also causes blood vessels to become porous, allowing the tissue to become edematous because proteins from the bloodstream leak into the extravascular space, which increases its osmolar load and draws water into the area.[3] Increased porosity of blood vessels also facilitates the entry of inflammatory cells like leukocytes into the wound site from the bloodstream.[17][18]
Polymorphonuclear neutrophils
Within an hour of wounding, polymorphonuclear neutrophils (PMNs) arrive at the wound site and become the predominant cells in the wound for the first two days after the injury occurs, with especially high numbers on the second day.[19] They are attracted to the site by fibronectin, growth factors, and substances such as kinins. Neutrophils phagocytise debris and kill bacteria by releasing free radicals in what is called a 'respiratory burst.[20][21] They also cleanse the wound by secreting proteases that break down damaged tissue. Functional neutrophils at the wound site only have life-spans of around 2 days, so they usually undergo apoptosis once they have completed their tasks and are engulfed and degraded by macrophages.[22]
Other leukocytes to enter the area include helper T cells, which secrete cytokines to cause more T cells to divide and to increase inflammation and enhance vasodilation and vessel permeability.[17][23] T cells also increase the activity of macrophages.[17]
Macrophages
One of the macrophage's roles is to phagocytize other expended phagocytes,[24] bacteria and damaged tissue,[19] and they also debride damaged tissue by releasing proteases.[25]
Macrophages function in regeneration[26][27] and are essential for wound healing.[19] They are stimulated by the low oxygen content of their surroundings to produce factors that induce and speed angiogenesis[20] and they also stimulate cells that reepithelialize the wound, create granulation tissue, and lay down a new extracellular matrix.[28] By secreting these factors, macrophages contribute to pushing the wound healing process into the next phase. They replace PMNs as the predominant cells in the wound by two days after injury.[24]
The spleen contains half the body's monocytes in reserve ready to be deployed to injured tissue.[29][30] Attracted to the wound site by growth factors released by platelets and other cells, monocytes from the bloodstream enter the area through blood vessel walls.[31] Numbers of monocytes in the wound peak one to one and a half days after the injury occurs.[23] Once they are in the wound site, monocytes mature into macrophages. Macrophages also secrete a number of factors such as growth factors and other cytokines, especially during the third and fourth post-wounding days. These factors attract cells involved in the proliferation stage of healing to the area.[15]
In wound healing that result in incomplete repair, scar contraction occurs, bringing varying gradations of structural imperfections, deformities and problems with flexibility.[32] Macrophages may restrain the contraction phase.[27] Scientists have reported that removing the macrophages from a salamander resulted in failure of a typical regeneration response (limb regeneration), instead bringing on a repair (scarring) response.[33][34]
Decline of inflammatory phase
As inflammation dies down, fewer inflammatory factors are secreted, existing ones are broken down, and numbers of neutrophils and macrophages are reduced at the wound site.[19] These changes indicate that the inflammatory phase is ending and the proliferative phase is underway.[19] In vitro evidence, obtained using the dermal equivalent model, suggests that the presence of macrophages actually delays wound contraction and thus the disappearance of macrophages from the wound may be essential for subsequent phases to occur.[27]
Because inflammation plays roles in fighting infection, clearing debris and inducing the proliferation phase, it is a necessary part of healing. However, inflammation can lead to tissue damage if it lasts too long.[7] Thus the reduction of inflammation is frequently a goal in therapeutic settings. Inflammation lasts as long as there is debris in the wound. Thus, if the individual's immune system is compromised and is unable to clear the debris from the wound and/or if excessive detritus, devitalized tissue, or microbial biofilm is present in the wound, these factors may cause a prolonged inflammatory phase and prevent the wound from properly commencing the proliferation phase of healing. This can lead to a chronic wound.
Proliferative phase
About two or three days after the wound occurs, fibroblasts begin to enter the wound site, marking the onset of the proliferative phase even before the inflammatory phase has ended.[35] As in the other phases of wound healing, steps in the proliferative phase do not occur in a series but rather partially overlap in time.
Angiogenesis
Also called neovascularization, the process of angiogenesis occurs concurrently with fibroblast proliferation when endothelial cells migrate to the area of the wound.[36] Because the activity of fibroblasts and epithelial cells requires oxygen and nutrients, angiogenesis is imperative for other stages in wound healing, like epidermal and fibroblast migration. The tissue in which angiogenesis has occurred typically looks red (is erythematous) due to the presence of capillaries.[36]
Angiogenesis occurs in overlapping phases in response to inflammation:
- Latent period: During the haemostatic and inflammatory phase of the wound healing process, vasodilation and permeabilisation allow leukocyte extravasation and phagocytic debridement and decontamination of the wound area. Tissue swelling aids later angiogenesis by expanding and loosening the existing collagenous extracellular matrix.
- Endothelial activation: As the wound macrophages switches from inflammatory to healing mode, it begins to secrete endothelial chemotactic and growth factors to attract adjacent endothelial cells. Activated endothelial cells respond by retracting and reducing cell junctions, loosening themselves from their embedded endothelium. Characteristically the activated endothelial cells show enlarged nucleoli.
- Degradation of endothelial basement membrane: The wound macrophages, mast cells and the endothelial cells themselves secrete proteases to break down existing vascular basal lamina.
- Vascular sprouting: With the breakdown of endothelial basement membrane, detached endothelial cells from pre-existing capillaries and post-capillary venules can divide and migrate chemotactically towards the wound, laying down new vessels in the process. Vascular sprouting can be aided by ambient hypoxia and acidosis in the wound environment, as hypoxia stimulates the endothelial transcription factor, hypoxia inducible factor (HIF) to transactivate angiogenic genes such as VEGF and GLUT1. Sprouted vessels can self-organise into luminal morphologies, and fusion of blind channels give rise to new capillary networks.
- Vascular maturation: the endothelium of vessels mature by laying down new endothelial extracellular matrix, followed by basal lamina formation. Lastly the vessel establishes a pericyte layer.
Stem cells of endothelial cells, originating from parts of uninjured blood vessels, develop pseudopodia and push through the ECM into the wound site to establish new blood vessels.[20]
Endothelial cells are attracted to the wound area by fibronectin found on the fibrin scab and chemotactically by angiogenic factors released by other cells,[37] e.g. from macrophages and platelets when in a low-oxygen environment. Endothelial growth and proliferation is also directly stimulated by hypoxia, and presence of lactic acid in the wound.[35] For example, hypoxia stimulates the endothelial transcription factor, hypoxia-inducible factor (HIF) to transactivate a set of proliferative genes including vascular endothelial growth factor (VEGF) and glucose transporter 1 (GLUT1).
To migrate, endothelial cells need collagenases and plasminogen activator to degrade the clot and part of the ECM.[3][19] Zinc-dependent metalloproteinases digest basement membrane and ECM to allow cell migration, proliferation and angiogenesis.[38]
When macrophages and other growth factor-producing cells are no longer in a hypoxic, lactic acid-filled environment, they stop producing angiogenic factors.[20] Thus, when tissue is adequately perfused, migration and proliferation of endothelial cells is reduced. Eventually blood vessels that are no longer needed die by apoptosis.[37]
Fibroplasia and granulation tissue formation
Simultaneously with angiogenesis, fibroblasts begin accumulating in the wound site. Fibroblasts begin entering the wound site two to five days after wounding as the inflammatory phase is ending, and their numbers peak at one to two weeks post-wounding.[19] By the end of the first week, fibroblasts are the main cells in the wound.[3] Fibroplasia ends two to four weeks after wounding.
As a model the mechanism of fibroplasia may be conceptualised as an analogous process to angiogenesis (see above) - only the cell type involved is fibroblasts rather than endothelial cells. Initially there is a latent phase where the wound undergoes plasma exudation, inflammatory decontamination and debridement. Oedema increases the wound histologic accessibility for later fibroplastic migration. Second, as inflammation nears completion, macrophage and mast cells release fibroblast growth and chemotactic factors to activate fibroblasts from adjacent tissue. Fibroblasts at this stage loosen themselves from surrounding cells and ECM. Phagocytes further release proteases that break down the ECM of neighbouring tissue, freeing the activated fibroblasts to proliferate and migrate towards the wound. The difference between vascular sprouting and fibroblast proliferation is that the former is enhanced by hypoxia, whilst the latter is inhibited by hypoxia. The deposited fibroblastic connective tissue matures by secreting ECM into the extracellular space, forming granulation tissue (see below). Lastly collagen is deposited into the ECM.
In the first two or three days after injury, fibroblasts mainly migrate and proliferate, while later, they are the main cells that lay down the collagen matrix in the wound site.[3] Origins of these fibroblasts are thought to be from the adjacent uninjured cutaneous tissue (although new evidence suggests that some are derived from blood-borne, circulating adult stem cells/precursors).[39] Initially fibroblasts utilize the fibrin cross-linking fibers (well-formed by the end of the inflammatory phase) to migrate across the wound, subsequently adhering to fibronectin.[37] Fibroblasts then deposit ground substance into the wound bed, and later collagen, which they can adhere to for migration.[15]
Granulation tissue functions as rudimentary tissue, and begins to appear in the wound already during the inflammatory phase, two to five days post wounding, and continues growing until the wound bed is covered. Granulation tissue consists of new blood vessels, fibroblasts, inflammatory cells, endothelial cells, myofibroblasts, and the components of a new, provisional extracellular matrix (ECM). The provisional ECM is different in composition from the ECM in normal tissue and its components originate from fibroblasts.[28] Such components include fibronectin, collagen, glycosaminoglycans, elastin, glycoproteins and proteoglycans.[37] Its main components are fibronectin and hyaluronan, which create a very hydrated matrix and facilitate cell migration.[31] Later this provisional matrix is replaced with an ECM that more closely resembles that found in non-injured tissue.
Growth factors (PDGF, TGF-β) and fibronectin encourage proliferation, migration to the wound bed, and production of ECM molecules by fibroblasts. Fibroblasts also secrete growth factors that attract epithelial cells to the wound site. Hypoxia also contributes to fibroblast proliferation and excretion of growth factors, though too little oxygen will inhibit their growth and deposition of ECM components, and can lead to excessive, fibrotic scarring.
Collagen deposition
One of fibroblasts' most important duties is the production of collagen.[36]
Collagen deposition is important because it increases the strength of the wound; before it is laid down, the only thing holding the wound closed is the fibrin-fibronectin clot, which does not provide much resistance to traumatic injury.[20] Also, cells involved in inflammation, angiogenesis, and connective tissue construction attach to, grow and differentiate on the collagen matrix laid down by fibroblasts.[40]
Type III collagen and fibronectin generally begin to be produced in appreciable amounts at somewhere between approximately 10 hours[41] and 3 days,[37] depending mainly on wound size. Their deposition peaks at one to three weeks.[28] They are the predominating tensile substances until the later phase of maturation, in which they are replaced by the stronger type I collagen.
Even as fibroblasts are producing new collagen, collagenases and other factors degrade it. Shortly after wounding, synthesis exceeds degradation so collagen levels in the wound rise, but later production and degradation become equal so there is no net collagen gain.[20] This homeostasis signals the onset of the later maturation phase. Granulation gradually ceases and fibroblasts decrease in number in the wound once their work is done.[42] At the end of the granulation phase, fibroblasts begin to commit apoptosis, converting granulation tissue from an environment rich in cells to one that consists mainly of collagen.[3]
Epithelialization
The formation of granulation tissue into an open wound allows the reepithelialization phase to take place, as epithelial cells migrate across the new tissue to form a barrier between the wound and the environment.[37] Basal keratinocytes from the wound edges and dermal appendages such as hair follicles, sweat glands and sebacious (oil) glands are the main cells responsible for the epithelialization phase of wound healing.[42] They advance in a sheet across the wound site and proliferate at its edges, ceasing movement when they meet in the middle. In healing that results in a scar, sweat glands, hair follicles[43][44] and nerves do not form. With the lack of hair follicles, nerves and sweat glands, the wound, and the resulting healing scar, provide a challenge to the body with regards to temperature control.[44]
Keratinocytes migrate without first proliferating.[45] Migration can begin as early as a few hours after wounding. However, epithelial cells require viable tissue to migrate across, so if the wound is deep it must first be filled with granulation tissue.[46] Thus the time of onset of migration is variable and may occur about one day after wounding.[47] Cells on the wound margins proliferate on the second and third day post-wounding in order to provide more cells for migration.[28]
If the basement membrane is not breached, epithelial cells are replaced within three days by division and upward migration of cells in the stratum basale in the same fashion that occurs in uninjured skin.[37] However, if the basement membrane is ruined at the wound site, reepithelization must occur from the wound margins and from skin appendages such as hair follicles and sweat and oil glands that enter the dermis that are lined with viable keratinocytes.[28] If the wound is very deep, skin appendages may also be ruined and migration can only occur from wound edges.[46]
Migration of keratinocytes over the wound site is stimulated by lack of contact inhibition and by chemicals such as nitric oxide.[48] Before they begin to migrate, cells must dissolve their desmosomes and hemidesmosomes, which normally anchor the cells by intermediate filaments in their cytoskeleton to other cells and to the ECM.[23] Transmembrane receptor proteins called integrins, which are made of glycoproteins and normally anchor the cell to the basement membrane by its cytoskeleton, are released from the cell's intermediate filaments and relocate to actin filaments to serve as attachments to the ECM for pseudopodia during migration.[23] Thus keratinocytes detach from the basement membrane and are able to enter the wound bed.[35]
Before they begin migrating, keratinocytes change shape, becoming longer and flatter and extending cellular processes like lamellipodia and wide processes that look like ruffles.[31] Actin filaments and pseudopodia form.[35] During migration, integrins on the pseudopod attach to the ECM, and the actin filaments in the projection pull the cell along.[23] The interaction with molecules in the ECM through integrins further promotes the formation of actin filaments, lamellipodia, and filopodia.[23]
Epithelial cells climb over one another in order to migrate.[42] This growing sheet of epithelial cells is often called the epithelial tongue.[45] The first cells to attach to the basement membrane form the stratum basale. These basal cells continue to migrate across the wound bed, and epithelial cells above them slide along as well.[45] The more quickly this migration occurs, the less of a scar there will be.[49]
Fibrin, collagen, and fibronectin in the ECM may further signal cells to divide and migrate. Like fibroblasts, migrating keratinocytes use the fibronectin cross-linked with fibrin that was deposited in inflammation as an attachment site to crawl across.[25][31][42]
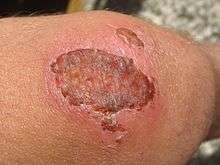
As keratinocytes migrate, they move over granulation tissue but stay underneath the scab, thereby separating the scab from the underlying tissue.[42][47] Epithelial cells have the ability to phagocytize debris such as dead tissue and bacterial matter that would otherwise obstruct their path. Because they must dissolve any scab that forms, keratinocyte migration is best enhanced by a moist environment, since a dry one leads to formation of a bigger, tougher scab.[25][37][42][50] To make their way along the tissue, keratinocytes must dissolve the clot, debris, and parts of the ECM in order to get through.[47][51] They secrete plasminogen activator, which activates plasminogen, turning it into plasmin to dissolve the scab. Cells can only migrate over living tissue,[42] so they must excrete collagenases and proteases like matrix metalloproteinases (MMPs) to dissolve damaged parts of the ECM in their way, particularly at the front of the migrating sheet.[47] Keratinocytes also dissolve the basement membrane, using instead the new ECM laid down by fibroblasts to crawl across.[23]
As keratinocytes continue migrating, new epithelial cells must be formed at the wound edges to replace them and to provide more cells for the advancing sheet.[25] Proliferation behind migrating keratinocytes normally begins a few days after wounding[46] and occurs at a rate that is 17 times higher in this stage of epithelialization than in normal tissues.[25] Until the entire wound area is resurfaced, the only epithelial cells to proliferate are at the wound edges.[45]
Growth factors, stimulated by integrins and MMPs, cause cells to proliferate at the wound edges. Keratinocytes themselves also produce and secrete factors, including growth factors and basement membrane proteins, which aid both in epithelialization and in other phases of healing.[52] Growth factors are also important for the innate immune defense of skin wounds by stimulation of the production of antimicrobial peptides and neutrophil chemotactic cytokines in keratinocytes.
Keratinocytes continue migrating across the wound bed until cells from either side meet in the middle, at which point contact inhibition causes them to stop migrating.[31] When they have finished migrating, the keratinocytes secrete the proteins that form the new basement membrane.[31] Cells reverse the morphological changes they underwent in order to begin migrating; they reestablish desmosomes and hemidesmosomes and become anchored once again to the basement membrane.[23] Basal cells begin to divide and differentiate in the same manner as they do in normal skin to reestablish the strata found in reepithelialized skin.[31]
Contraction
Contraction is a key phase of wound healing with repair. If contraction continues for too long, it can lead to disfigurement and loss of function.[32] Thus there is a great interest in understanding the biology of wound contraction, which can be modelled in vitro using the collagen gel contraction assay or the dermal equivalent model.[27][53]
Contraction commences approximately a week after wounding, when fibroblasts have differentiated into myofibroblasts.[54] In full thickness wounds, contraction peaks at 5 to 15 days post wounding.[37] Contraction can last for several weeks[46] and continues even after the wound is completely reepithelialized.[3] A large wound can become 40 to 80% smaller after contraction.[31][42] Wounds can contract at a speed of up to 0.75 mm per day, depending on how loose the tissue in the wounded area is.[37] Contraction usually does not occur symmetrically; rather most wounds have an 'axis of contraction' which allows for greater organization and alignment of cells with collagen.[54]
At first, contraction occurs without myofibroblast involvement.[55] Later, fibroblasts, stimulated by growth factors, differentiate into myofibroblasts. Myofibroblasts, which are similar to smooth muscle cells, are responsible for contraction.[55] Myofibroblasts contain the same kind of actin as that found in smooth muscle cells.[32]
Myofibroblasts are attracted by fibronectin and growth factors and they move along fibronectin linked to fibrin in the provisional ECM in order to reach the wound edges.[25] They form connections to the ECM at the wound edges, and they attach to each other and to the wound edges by desmosomes. Also, at an adhesion called the fibronexus, actin in the myofibroblast is linked across the cell membrane to molecules in the extracellular matrix like fibronectin and collagen.[55] Myofibroblasts have many such adhesions, which allow them to pull the ECM when they contract, reducing the wound size.[32] In this part of contraction, closure occurs more quickly than in the first, myofibroblast-independent part.[55]
As the actin in myofibroblasts contracts, the wound edges are pulled together. Fibroblasts lay down collagen to reinforce the wound as myofibroblasts contract.[3] The contraction stage in proliferation ends as myofibroblasts stop contracting and commit apoptosis.[32] The breakdown of the provisional matrix leads to a decrease in hyaluronic acid and an increase in chondroitin sulfate, which gradually triggers fibroblasts to stop migrating and proliferating.[19] These events signal the onset of the maturation stage of wound healing.
Maturation and remodeling
When the levels of collagen production and degradation equalize, the maturation phase of tissue repair is said to have begun.[20] During maturation, type III collagen, which is prevalent during proliferation, is replaced by type I collagen.[17] Originally disorganized collagen fibers are rearranged, cross-linked, and aligned along tension lines.[31] The onset of the maturation phase may vary extensively, depending on the size of the wound and whether it was initially closed or left open,[28] ranging from approximately 3 days[41] to 3 weeks.[56] The maturation phase can last for a year or longer, similarly depending on wound type.[28]
As the phase progresses, the tensile strength of the wound increases.[28] Collagen will reach approximately 20% of its tensile strength after 3 weeks, increasing to 80% by 12th week. The maximum scar strength is 80% of that of unwounded skin.[57] Since activity at the wound site is reduced, the scar loses its red appearance as blood vessels that are no longer needed are removed by apoptosis.[20]
The phases of wound healing normally progress in a predictable, timely manner; if they do not, healing may progress inappropriately to either a chronic wound[7] such as a venous ulcer or pathological scarring such as a keloid scar.[58][59]
Factors affecting wound healing
Many factors controlling the efficacy, speed, and manner of wound healing fall under two types: local and systemic factors.[2]
Local factors
- Moisture; keeping a wound moist rather than dry makes wound healing more rapid and with less pain and less scarring[60]
- Mechanical factors
- Oedema
- Ionizing radiation
- Faculty technique of wound closure
- Ischemia and necrosis
- Foreign bodies. Sharp, small foreign bodies can penetrate the skin leaving little surface wound but causing internal injury and internal bleeding. For a glass foreign body, "frequently, an innocent skin wound disguises the extensive nature of the injuries beneath".[61] First-degree nerve injury requires a few hours to a few weeks to recover.[62] If a foreign body passes by a nerve and causes first-degree nerve injury during entry, then the sensation of the foreign body or pain due to internal wounding may be delayed by a few hours to a few weeks after entry. A sudden increase in pain during the first few weeks of wound healing could be a sign of a recovered nerve reporting internal injuries rather than a newly developed infection.
- Low oxygen tension
- Perfusion
Systemic factors
- Inflammation
- Diabetes – Individuals with diabetes demonstrate reduced capability in the healing of acute wounds. Additionally, diabetic individuals are susceptible to developing chronic diabetic foot ulcers, a serious complication of diabetes which affects 15% of people with diabetes and accounts for 84% of all diabetes-related lower leg amputations.[63] The impaired healing abilities of diabetics with diabetic foot ulcers and/or acute wounds involves multiple pathophysiological mechanisms.[64] This impaired healing involves hypoxia, fibroblast and epidermal cell dysfunction, impaired angiogenesis and neovascularization, high levels of metalloproteases, damage from reactive oxygen species and AGEs (advanced glycation end-products), decreased host immune resistance, and neuropathy.[64]
- Nutrients – Malnutrition or nutritional deficiencies have a recognizable impact on wound healing post trauma or surgical intervention.[65] Nutrients including proteins, carbohydrates, arginine, glutamine, polyunsaturated fatty acids, vitamin A, vitamin C, vitamin E, magnesium, copper, zinc and iron all play significant roles in wound healing.[64] Fats and carbohydrates provide the majority of energy required for wound healing. Glucose is the most prominent source of fuel and it is used to create cellular ATP, providing energy for angiogenesis and the deposition of new tissues.[64] As the nutritional needs of each patient and their associated wound are complex, it is suggested that tailored nutritional support would benefit both acute and chronic wound healing.[64]
- Metabolic diseases
- Immunosuppression
- Connective tissue disorders
- Smoking – Smoking causes a delay in the speed of wound repair notably in the proliferative and inflammatory phases. It also increases the likelihood of certain complications such as wound rupture, wound and flap necrosis, decrease in wound tensile strength and infection.[64] Passive smoking also impairs a proper wound healing process.[66]
- Age – Increased age (over 60 years) is a risk factor for impaired wound healing.[64] It is recognized that, in older adults of otherwise overall good health, the effects of aging causes a temporal delay in healing, but no major impairment with regard to the quality of healing.[67] Delayed wound healing in patients of increasing age is associated with altered inflammatory response; for example delayed T-cell infiltration of the wound with alterations in the production of chemokines, and reduced macrophage phagocytic capacity.[68]
- Alcohol – Alcohol consumption impairs wound healing and also increases the chances of infection. Alcohol affects the proliferative phase of healing. A single unit of alcohol causes a negative effect on re-epithelialization, wound closure, collagen production and angiogenesis.[64]
Research and development
Up until about 2000, the classic paradigm of wound healing, involving stem cells restricted to organ-specific lineages, had never been seriously challenged. Since then, the notion of adult stem cells having cellular plasticity or the ability to differentiate into non-lineage cells has emerged as an alternative explanation.[1] To be more specific, hematopoietic progenitor cells (that give rise to mature cells in the blood) may have the ability de-differentiate back into hematopoietic stem cells and/or transdifferentiate into non-lineage cells, such as fibroblasts.[39]
Stem cells and cellular plasticity
Multipotent adult stem cells have the capacity to be self-renewing and give rise to different cell types. Stem cells give rise to progenitor cells, which are cells that are not self-renewing, but can generate several types of cells. The extent of stem cell involvement in cutaneous (skin) wound healing is complex and not fully understood.
It is thought that the epidermis and dermis are reconstituted by mitotically active stem cells that reside at the apex of rete ridges (basal stem cells or BSC), the bulge of hair follicles (hair follicular stem cell or HFSC), and the papillary dermis (dermal stem cells).[1] Moreover, bone marrow may also contain stem cells that play a major role in cutaneous wound healing.[39]
In rare circumstances, such as extensive cutaneous injury, self-renewal subpopulations in the bone marrow are induced to participate in the healing process, whereby they give rise to collagen-secreting cells that seem to play a role during wound repair.[1] These two self-renewal subpopulations are (1) bone marrow-derived mesenchymal stem cells (MSC) and (2) hematopoietic stem cells (HSC). Bone marrow also harbors a progenitor subpopulation (endothelial progenitor cells or EPC) that, in the same type of setting, are mobilized to aid in the reconstruction of blood vessels.[39] Moreover, it thought that, extensive injury to skin also promotes the early trafficking of a unique subclass of leukocytes (circulating fibrocytes) to the injured region, where they perform various functions related to wound healing.[1]
Wound repair versus regeneration
An injury is an interruption of morphology and/or functionality of a given tissue. After injury, structural tissue heals with incomplete or complete regeneration.[69][70] Tissue without an interruption to the morphology almost always completely regenerates. An example of complete regeneration without an interruption of the morphology is non-injured tissue, such as skin.[71] Non-injured skin has a continued replacement and regeneration of cells which always results in complete regeneration.[71]
There is a subtle distinction between 'repair' and 'regeneration'.[1][69][70] Repair means incomplete regeneration.[69] Repair or incomplete regeneration, refers to the physiologic adaptation of an organ after injury in an effort to re-establish continuity without regards to exact replacement of lost/damaged tissue.[69] True tissue regeneration or complete regeneration,[70] refers to the replacement of lost/damaged tissue with an ‘exact’ copy, such that both morphology and functionality are completely restored.[70] Though after injury mammals can completely regenerate spontaneously, they usually do not completely regenerate. An example of a tissue regenerating completely after an interruption of morphology is the endometrium; the endometrium after the process of breakdown via the menstruation cycle heals with complete regeneration.[71]
In some instances, after a tissue breakdown, such as in skin, a regeneration closer to complete regeneration may be induced by the use of biodegradable (collagen-glycoaminoglycan) scaffolds. These scaffolds are structurally analogous to extracellular matrix (ECM) found in normal/un-injured dermis.[72] Fundamental conditions required for tissue regeneration often oppose conditions that favor efficient wound repair, including inhibition of (1) platelet activation, (2) inflammatory response, and (3) wound contraction.[1] In addition to providing support for fibroblast and endothelial cell attachment, biodegradable scaffolds inhibit wound contraction, thereby allowing the healing process to proceed towards a more-regenerative/less-scarring pathway. Pharmaceutical agents have been investigated which may be able to turn off myofibroblast differentiation.[73]
A new way of thinking derived from the notion that heparan sulfates are key player in tissue homeostasis: the process that makes the tissue replace dead cells by identical cells. In wound areas, tissue homeostasis is lost as the heparan sulfates are degraded preventing the replacement of dead cells by identical cells. Heparan sulfate analogues cannot be degraded by all know heparanases and glycanases and bind to the free heparin sulfate binding spots on the ECM, therefore preserving the normal tissue homeostasis and preventing scarring.[74][75][76]
Repair or regeneration with regards to hypoxia-inducible factor 1-alpha (HIF-1a). In normal circumstances after injury HIF-1a is degraded by prolyl hydroxylases (PHDs). Scientists found that the simple up-regulation of HIF-1a via PHD inhibitors regenerates lost or damaged tissue in mammals that have a repair response; and the continued down-regulation of Hif-1a results in healing with a scarring response in mammals with a previous regenerative response to the loss of tissue. The act of regulating HIF-1a can either turn off, or turn on the key process of mammalian regeneration.[77][78]
Scarless wound healing
Scarless wound healing is a concept based on the healing or repair of the skin (or other tissue/organs) after injury with the aim of healing with subjectively and relatively less scar tissue than normally expected. Scarless healing is sometimes mixed up with the concept of scar free healing, which is wound healing which results in absolutely no scar (free of scarring). However they are different concepts.
A reverse to scarless wound healing is scarification (wound healing to scar more). Historically, certain cultures consider scarification attractive;[79] however, this is generally not the case in the modern western society, in which many patients are turning to plastic surgery clinics with unrealistic expectations. Depending on scar type, treatment may be invasive (intralesional steroid injections, surgery) and/or conservative (compression therapy, topical silicone gel, brachytherapy, photodynamic therapy).[80] Clinical judgment is necessary to successfully balance the potential benefits of the various treatments available against the likelihood of a poor response and possible complications resulting from these treatments. Many of these treatments may only have a placebo effect, and the evidence base for the use of many current treatments is poor.[81]
Since the 1960s, comprehension of the basic biologic processes involved in wound repair and tissue regeneration have expanded due to advances in cellular and molecular biology.[82] Currently, the principal goals in wound management are to achieve rapid wound closure with a functional tissue that has minimal aesthetic scarring.[83] However, the ultimate goal of wound healing biology is to induce a more perfect reconstruction of the wound area. Scarless wound healing only occurs in mammalian foetal tissues[84] and complete regeneration is limited to lower vertebrates, such as salamanders, and invertebrates.[85] In adult humans, injured tissue are repaired by collagen deposition, collagen remodelling and eventual scar formation, where fetal wound healing is believed to be more of a regenerative process with minimal or no scar formation.[84] Therefore, foetal wound healing can be used to provide an accessible mammalian model of an optimal healing response in adult human tissues. Clues as to how this might be achieved come from studies of wound healing in embryos, where repair is fast and efficient and results in essentially perfect regeneration of any lost tissue.
The etymology of the term scarless wound healing has a long history.[86][87][88] In print the antiquated concept of scarless healing was brought up the early 20th century and appeared in a paper published in the London Lancet. This process involved cutting in a surgical slant, instead of a right angle…; it was described in various Newspapers.[86][87][88]
Cancer
After inflammation, restoration of normal tissue integrity and function is preserved by feedback interactions between diverse cell types mediated by adhesion molecules and secreted cytokines. Disruption of normal feedback mechanisms in cancer threatens tissue integrity and enables a malignant tumor to escape the immune system.[89][90] An example of the importance of the wound healing response within tumors is illustrated in work by Howard Chang and colleagues at Stanford University studying Breast cancers.[8]
Simulating wound healing from a growth perspective
Considerable effort has been devoted to understanding the physical relationships governing wound healing and subsequent scarring, with mathematical models and simulations developed to elucidate these relationships.[91] The growth of tissue around the wound site is a result of the migration of cells and collagen deposition by these cells. The alignment of collagen describes the degree of scarring; basket-weave orientation of collagen is characteristic of normal skin, whereas aligned collagen fibers lead to significant scarring.[92] It has been shown that the growth of tissue and extent of scar formation can be controlled by modulating the stress at a wound site.[93]
The growth of tissue can be simulated using the aforementioned relationships from a biochemical and biomechanical point of view. The biologically active chemicals that play an important role in wound healing are modeled with Fickian diffusion to generate concentration profiles. The balance equation for open systems when modeling wound healing incorporates mass growth due to cell migration and proliferation. Here the following equation is used:
Dtρ0 = Div (R) + R0,
where ρ represents mass density, R represents a mass flux (from cell migration), and R0 represents a mass source (from cell proliferation, division, or enlargement).[94] Relationships like these can be incorporated into an agent-based models, where the sensitivity to single parameters such as initial collagen alignment, cytokine properties, and cell proliferation rates can be tested.[95]
Wound closure intentions
Successful wound healing is dependent on various cell types, molecular mediators and structural elements.[96]
Primary intention
Primary intention is the healing of a clean wound without tissue loss.[96] In this process, wound edges are brought together, so that they are adjacent to each other (re-approximated). Wound closure is performed with sutures (stitches), staples, or adhesive tape or glue.
Primary intention can only be implemented when the wound is precise and there is minimal disruption to the local tissue and the epithelial basement membrane, e.g. surgical incisions.[97]
This process is faster than healing by secondary intention.[96] There is also less scarring associated with primary intention, as there are no large tissue losses to be filled with granulation tissue.[96] (Primary intention does require some granulation tissue to form.)
- Examples of primary intention include: well-repaired lacerations, well reduced bone fractures, healing after flap surgery.
- Early removal of dressings from clean or clean-contaminated wounds does affect primary healing of wounds.[98]
Secondary intention
- Secondary intention is implemented when primary intention is not possible.
- This is due to wounds being created by major trauma in which there has been a significant loss in tissue or tissue damage.[97]
- The wound is allowed to granulate.
- Surgeon may pack the wound with a gauze or use a drainage system.
- Granulation results in a broader scar.
- Healing process can be slow due to presence of drainage from infection.
- Wound care must be performed daily to encourage wound debris removal to allow for granulation tissue formation.
- Using antibiotics or antiseptics for the surgical wound healing by secondary intention is controversial.[99]
- Examples: gingivectomy, gingivoplasty, tooth extraction sockets, poorly reduced fractures, burns, severe lacerations, pressure ulcers.
- There is insufficient evidence that the choice of dressings or topical agents affects the secondary healing of wounds.[100]
- There is lack of evidence for the effectiveness of negative pressure wound therapy in wound healing by secondary intention.[101]
Tertiary intention
(Delayed primary closure or secondary suture):
- The wound is initially cleaned, debrided and observed, typically 4 or 5 days before closure.
- The wound is purposely left open.
- Examples: healing of wounds by use of tissue grafts.
If the wound edges are not reapproximated immediately, delayed primary wound healing transpires. This type of healing may be desired in the case of contaminated wounds. By the fourth day, phagocytosis of contaminated tissues is well underway, and the processes of epithelization, collagen deposition, and maturation are occurring. Foreign materials are walled off by macrophages that may metamorphose into epithelioid cells, which are encircled by mononuclear leukocytes, forming granulomas. Usually the wound is closed surgically at this juncture, and if the "cleansing" of the wound is incomplete, chronic inflammation can ensue, resulting in prominent scarring.
Overview of involved growth factors
Following are the main growth factors involved in wound healing:
| Growth factor | Abbreviation | Main origins | Effects |
|---|---|---|---|
| Epidermal growth factor | EGF |
|
|
| Transforming growth factor-α | TGF-α |
|
|
| Hepatocyte growth factor | HGF |
|
|
| Vascular endothelial growth factor | VEGF |
|
|
| Platelet derived growth factor | PDGF |
|
|
| Fibroblast growth factor 1 and 2 | FGF-1, −2 |
|
|
| Transforming growth factor-β | TGF-β |
|
|
| Keratinocyte growth factor | KGF |
|
|
| Unless else specified in boxes, then reference is:[102] | |||
Complications of wound healing
The major complications are many:
- Deficient scar formation: Results in wound dehiscence or rupture of the wound due to inadequate formation of granulation tissue.
- Excessive scar formation: Hypertrophic scar, keloid, desmoid.
- Exuberant granulation (proud flesh).
- Deficient contraction (in skin grafts) or excessive contraction (in burns).
- Others: Dystrophic calcification, pigmentary changes, painful scars, incisional hernia
Other complications can include Infection and Marjolin's ulcer.
Biologics, skin substitutes, biomembranes and scaffolds
Advancements in the clinical understanding of wounds and their pathophysiology have commanded significant biomedical innovations in the treatment of acute, chronic, and other types of wounds. Many biologics, skin substitutes, biomembranes and scaffolds have been developed to facilitate wound healing through various mechanisms.[103] This includes a number of products under the trade names such as Epicel, Laserskin, Transcyte, Dermagraft, AlloDerm/Strattice, Biobrane, Integra, Apligraf, OrCel, GraftJacket and PermaDerm.[104]
See also
Notes and references
- Nguyen DT, Orgill DP, Murphy GT (2009). "4 The Pathophysiologic Basis for Wound Healing and Cutaneous Regeneration". Biomaterials for Treating Skin Loss. Elsevier. pp. 25–57. Orgill DP, Blanco C (editors). ISBN 978-1-84569-554-5.
- Rieger S, Zhao H, Martin P, Abe K, Lisse TS (January 2015). "The role of nuclear hormone receptors in cutaneous wound repair". Cell Biochemistry and Function. 33 (1): 1–13. doi:10.1002/cbf.3086. PMC 4357276. PMID 25529612.
- Stadelmann WK, Digenis AG, Tobin GR (August 1998). "Physiology and healing dynamics of chronic cutaneous wounds". American Journal of Surgery. 176 (2A Suppl): 26S–38S. doi:10.1016/S0002-9610(98)00183-4. PMID 9777970.
- Enoch, S. Price, P. (2004). Cellular, molecular and biochemical differences in the pathophysiology of healing between acute wounds, chronic wounds and wounds in the elderly Archived 2017-07-06 at the Wayback Machine.
- Rasche H (2001). "Haemostasis and thrombosis: an overview". European Heart Journal Supplements. 3 (Supplement Q): Q3–Q7. doi:10.1016/S1520-765X(01)90034-3.
- Versteeg HH, Heemskerk JW, Levi M, Reitsma PH (January 2013). "New fundamentals in hemostasis". Physiological Reviews. 93 (1): 327–58. doi:10.1152/physrev.00016.2011. PMID 23303912.
- Midwood KS, Williams LV, Schwarzbauer JE (June 2004). "Tissue repair and the dynamics of the extracellular matrix". The International Journal of Biochemistry & Cell Biology. 36 (6): 1031–7. doi:10.1016/j.biocel.2003.12.003. PMID 15094118.
- Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, et al. (February 2004). "Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds". PLoS Biology. 2 (2): E7. doi:10.1371/journal.pbio.0020007. PMC 314300. PMID 14737219.
- Garg, H.G. (2000). Scarless Wound Healing. New York Marcel Dekker, Inc. Electronic book.
- Reference list is found on image main page.
- Cubison TC, Pape SA, Parkhouse N (December 2006). "Evidence for the link between healing time and the development of hypertrophic scars (HTS) in paediatric burns due to scald injury". Burns. 32 (8): 992–9. doi:10.1016/j.burns.2006.02.007. PMID 16901651.
- Kraft J, Lynde C. "Giving Burns the First, Second and Third Degree - Classification of burns". skincareguide.ca. Archived from the original on 26 December 2011. Retrieved 31 January 2012.
Formation of a thick eschar, slow healing (>1month), Obvious scarring,
- "POST BURN SCAR RELATIVE TO RE-EPITHELIALIZATION". Burnsurgery.org. 2011. Archived from the original on 25 April 2012. Retrieved 16 March 2011.
Healing in 2 weeks – minimal to no scar; Healing in 3 weeks – minimal to no scar except in high risk scar formers;Healing in 4 weeks or more – hypertrophic in more than 50% of patients
- Galko MJ, Krasnow MA (August 2004). "Cellular and genetic analysis of wound healing in Drosophila larvae". PLoS Biology. 2 (8): E239. doi:10.1371/journal.pbio.0020239. PMC 479041. PMID 15269788.
- Rosenberg L., de la Torre J. (2006). Wound Healing, Growth Factors Archived 2008-11-21 at the Wayback Machine. Emedicine.com. Accessed January 20, 2008.
- Sandeman SR, Allen MC, Liu C, Faragher RG, Lloyd AW (November 2000). "Human keratocyte migration into collagen gels declines with in vitro ageing". Mechanisms of Ageing and Development. 119 (3): 149–57. doi:10.1016/S0047-6374(00)00177-9. PMID 11080534.
- Dealey C. (1999). The care of wounds: A guide for nurses. Oxford; Malden, Mass. Blackwell Science. Electronic book.
- Theoret CL (2004). "Update on wound repair". Clinical Techniques in Equine Practice. 3 (2): 110–122. doi:10.1053/j.ctep.2004.08.009.
- de la Torre J., Sholar A. (2006). Wound healing: Chronic wounds Archived 2008-10-29 at the Wayback Machine. Emedicine.com. Accessed January 20, 2008.
- Greenhalgh DG (September 1998). "The role of apoptosis in wound healing". The International Journal of Biochemistry & Cell Biology. 30 (9): 1019–30. doi:10.1016/S1357-2725(98)00058-2. PMID 9785465.
- Muller MJ, Hollyoak MA, Moaveni Z, Brown TL, Herndon DN, Heggers JP (December 2003). "Retardation of wound healing by silver sulfadiazine is reversed by Aloe vera and nystatin". Burns. 29 (8): 834–6. doi:10.1016/S0305-4179(03)00198-0. PMID 14636760.
- Martin P, Leibovich SJ (November 2005). "Inflammatory cells during wound repair: the good, the bad and the ugly". Trends in Cell Biology. 15 (11): 599–607. doi:10.1016/j.tcb.2005.09.002. PMID 16202600.
- Santoro MM, Gaudino G (March 2005). "Cellular and molecular facets of keratinocyte reepithelization during wound healing". Experimental Cell Research. 304 (1): 274–86. doi:10.1016/j.yexcr.2004.10.033. PMID 15707592.
- "The phases of cutaneous wound healing" (PDF). Expert Reviews in Molecular Medicine. Cambridge University Press. 5. 21 March 2003. Archived from the original (PDF) on 8 March 2008.
- Deodhar AK, Rana RE (1997). "Surgical physiology of wound healing: a review". Journal of Postgraduate Medicine. 43 (2): 52–6. PMID 10740722. Archived from the original on 2011-02-26. Retrieved 2005-10-27.
- Ovchinnikov DA (September 2008). "Macrophages in the embryo and beyond: much more than just giant phagocytes". Genesis. researchgate.net. 46 (9): 447–62. doi:10.1002/dvg.20417. PMID 18781633.
Macrophages are present essentially in all tissues, beginning with embryonic development and, in addition to their role in host defense and in the clearance of apoptotic cells, are being increasingly recognized for their trophic function and role in regeneration.
- Newton PM, Watson JA, Wolowacz RG, Wood EJ (August 2004). "Macrophages restrain contraction of an in vitro wound healing model". Inflammation. 28 (4): 207–14. doi:10.1023/B:IFLA.0000049045.41784.59. PMID 15673162.
- Mercandetti M, Cohen AJ (2005). "Wound Healing: Healing and Repair". Emedicine.com. Archived from the original on 21 November 2008. Retrieved 20 January 2008.
- Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, et al. (July 2009). "Identification of splenic reservoir monocytes and their deployment to inflammatory sites". Science. 325 (5940): 612–6. Bibcode:2009Sci...325..612S. doi:10.1126/science.1175202. PMC 2803111. PMID 19644120.
- Jia T, Pamer EG (July 2009). "Immunology. Dispensable but not irrelevant". Science. 325 (5940): 549–50. Bibcode:2009Sci...325..549J. doi:10.1126/science.1178329. PMC 2917045. PMID 19644100.
- Lorenz HP, Longaker MT (2003). "Wounds: Biology, Pathology, and Management" (PDF). In Norton JA (ed.). Surgery. New York, NY: Springer. pp. 191–208. doi:10.1007/978-0-387-68113-9_10. ISBN 978-0-387-30800-5. Archived from the original (PDF) on 24 August 2014.
- Hinz B (April 2006). "Masters and servants of the force: the role of matrix adhesions in myofibroblast force perception and transmission". European Journal of Cell Biology. 85 (3–4): 175–81. doi:10.1016/j.ejcb.2005.09.004. PMID 16546559.
- Souppouris, Aaron (2013-05-23). "Scientists identify cell that could hold the secret to limb regeneration". the verge.com. Archived from the original on 2017-07-31. Retrieved 2017-09-18.
Researchers have identified a cell that aids limb regrowth in Salamanders. Macrophages are a type of repairing cell that devour dead cells and pathogens, and trigger other immune cells to respond to pathogens.
- Godwin JW, Pinto AR, Rosenthal NA (June 2013). "Macrophages are required for adult salamander limb regeneration". Proceedings of the National Academy of Sciences of the United States of America. University of Texas. 110 (23): 9415–20. Bibcode:2013PNAS..110.9415G. doi:10.1073/pnas.1300290110. PMC 3677454. PMID 23690624.
- Falanga V. (2005). Wound Healing. American Academy of Dermatology (AAD).
- Kuwahara R.T. and Rasberry R. 2007. Chemical Peels Archived 2008-10-25 at the Wayback Machine. Emedicine.com. Accessed September 15, 2007.
- Romo T. and Pearson J.M. 2005. Wound Healing, Skin Archived 2008-12-07 at the Wayback Machine. Emedicine.com. Accessed December 27, 2006.
- Lansdown AB, Sampson B, Rowe A (February 2001). "Experimental observations in the rat on the influence of cadmium on skin wound repair". International Journal of Experimental Pathology. 82 (1): 35–41. doi:10.1046/j.1365-2613.2001.00180.x. PMC 2517695. PMID 11422539.
- Song G, Nguyen DT, Pietramaggiori G, Scherer S, Chen B, Zhan Q, Ogawa R, Yannas IV, Wagers AJ, Orgill DP, Murphy GF (2010). "Use of the parabiotic model in studies of cutaneous wound healing to define the participation of circulating cells". Wound Repair and Regeneration. 18 (4): 426–32. doi:10.1111/j.1524-475X.2010.00595.x. PMC 2935287. PMID 20546556.
- Ruszczak Z (November 2003). "Effect of collagen matrices on dermal wound healing". Advanced Drug Delivery Reviews. 55 (12): 1595–611. doi:10.1016/j.addr.2003.08.003. PMID 14623403.
- Fig. 9-1. The cellular, biochemical, and mechanical phases of wound healing. Pollock RE, Brunicardi FC, Andersen DK, Billiar TR, Dunn D, Hunter JG, Matthews JJ (2009). Schwartz's Principles of Surgery, Ninth Edition. McGraw-Hill Professional. ISBN 978-0-07-154769-7.
- DiPietro LA, Burns AL, eds. (2003). Wound Healing: Methods and Protocols. Methods in Molecular Medicine. Totowa, N.J.: Humana Press.
- Fu XB, Sun TZ, Li XK, Sheng ZY (February 2005). "Morphological and distribution characteristics of sweat glands in hypertrophic scar and their possible effects on sweat gland regeneration". Chinese Medical Journal. 118 (3): 186–91. PMID 15740645. Archived from the original on 2018-06-20. Retrieved 2014-06-18.
- "BURN INJURIES". nationaltraumainstitute.org. Archived from the original on 3 March 2016. Retrieved 13 July 2016.
When the dermis is destroyed, the scars do not regrow hair, nerves or sweat glands, providing additional challenges to body temperature control.
- Bartkova J, Grøn B, Dabelsteen E, Bartek J (February 2003). "Cell-cycle regulatory proteins in human wound healing". Archives of Oral Biology. 48 (2): 125–32. doi:10.1016/S0003-9969(02)00202-9. PMID 12642231.
- Mulvaney M. and Harrington A. 1994. Chapter 7: Cutaneous trauma and its treatment. In, Textbook of Military Medicine: Military Dermatology. Office of the Surgeon General, Department of the Army. Virtual Naval Hospital Project. Accessed through web archive on September 15, 2007.
- Larjava H., Koivisto L., and Hakkinen L. 2002. Chapter 3: Keratinocyte Interactions with Fibronectin During Wound Healing. In, Heino, J. and Kahari, V.M. Cell Invasion. Medical Intelligence Unit; 33. Georgetown, Tex., Austin, Tex Landes Bioscience, Inc. Electronic book.
- Witte MB, Barbul A (April 2002). "Role of nitric oxide in wound repair". American Journal of Surgery. 183 (4): 406–12. doi:10.1016/S0002-9610(02)00815-2. PMID 11975928.
- Son HJ, Bae HC, Kim HJ, Lee DH, Han D, Park J (2005). "Effects of β-glucan on proliferation and migration of fibroblasts". Current Applied Physics. 5 (5): 468–71. Bibcode:2005CAP.....5..468S. doi:10.1016/j.cap.2005.01.011.
- Falanga V (2004). "The chronic wound: impaired healing and solutions in the context of wound bed preparation". Blood Cells, Molecules & Diseases. 32 (1): 88–94. doi:10.1016/j.bcmd.2003.09.020. PMID 14757419.
- Etscheid M, Beer N, Dodt J (December 2005). "The hyaluronan-binding protease upregulates ERK1/2 and PI3K/Akt signalling pathways in fibroblasts and stimulates cell proliferation and migration". Cellular Signalling. 17 (12): 1486–94. doi:10.1016/j.cellsig.2005.03.007. PMID 16153533.
- Bayram Y, Deveci M, Imirzalioglu N, Soysal Y, Sengezer M (October 2005). "The cell based dressing with living allogenic keratinocytes in the treatment of foot ulcers: a case study". British Journal of Plastic Surgery. 58 (7): 988–96. doi:10.1016/j.bjps.2005.04.031. PMID 16040019.
- Grinnell F (February 1994). "Fibroblasts, myofibroblasts, and wound contraction". The Journal of Cell Biology. 124 (4): 401–4. doi:10.1083/jcb.124.4.401. PMC 2119916. PMID 8106541.
- Eichler MJ, Carlson MA (February 2006). "Modeling dermal granulation tissue with the linear fibroblast-populated collagen matrix: a comparison with the round matrix model". Journal of Dermatological Science. 41 (2): 97–108. doi:10.1016/j.jdermsci.2005.09.002. PMID 16226016.
- Mirastschijski U, Haaksma CJ, Tomasek JJ, Agren MS (October 2004). "Matrix metalloproteinase inhibitor GM 6001 attenuates keratinocyte migration, contraction and myofibroblast formation in skin wounds". Experimental Cell Research. 299 (2): 465–75. doi:10.1016/j.yexcr.2004.06.007. PMID 15350544.
- worldwidewounds.com Archived 2011-07-05 at the Wayback Machine > Figure 3 – The time relationship between the different processes of wound healing. Archived 2011-07-18 at the Wayback Machine by Gregory S Schultz, Glenn Ladwig and Annette Wysocki – in turn adapted from Asmussen PD, Sollner B. Mechanism of wound healing. In: Wound Care. Tutorial Medical Series. Stuttgart: Hippokrates Verlag, 1993.
- Morton LM, Phillips TJ (April 2016). "Wound healing and treating wounds: Differential diagnosis and evaluation of chronic wounds". Journal of the American Academy of Dermatology. 74 (4): 589–605, quiz 605–6. doi:10.1016/j.jaad.2015.08.068. PMID 26979352.
- O'Leary R, Wood EJ, Guillou PJ (2002). "Pathological scarring: strategic interventions". The European Journal of Surgery = Acta Chirurgica. 168 (10): 523–34. PMID 12666691.
- Desmoulière A, Chaponnier C, Gabbiani G (2005). "Tissue repair, contraction, and the myofibroblast". Wound Repair and Regeneration. 13 (1): 7–12. doi:10.1111/j.1067-1927.2005.130102.x. PMID 15659031.
- Metzger S (September 2004). "Clinical and financial advantages of moist wound management". Home Healthcare Nurse. 22 (9): 586–90. doi:10.1097/00004045-200409000-00003. PMID 15359168.
- Iconomou TG, Zuker RM, Michelow BJ (1993). "Management of major penetrating glass injuries to the upper extremities in children and adolescents". Microsurgery. 14 (2): 91–6. doi:10.1002/micr.1920140202. PMID 8469109.
- "Nerve injury". Johns Hopkins Medicine. The Johns Hopkins University, The Johns Hopkins Hospital, and Johns Hopkins Health System. Archived from the original on 27 September 2016. Retrieved 2 October 2016.
- Brem H, Tomic-Canic M (May 2007). "Cellular and molecular basis of wound healing in diabetes". The Journal of Clinical Investigation. 117 (5): 1219–22. doi:10.1172/jci32169. PMC 1857239. PMID 17476353.
- Guo S, Dipietro LA (March 2010). "Factors affecting wound healing". Journal of Dental Research. 89 (3): 219–29. doi:10.1177/0022034509359125. PMC 2903966. PMID 20139336.
- Arnold M, Barbul A (June 2006). "Nutrition and wound healing". Plastic and Reconstructive Surgery. 117 (7 Suppl): 42S–58S. doi:10.1097/01.prs.0000225432.17501.6c. PMID 16799374.
- Wong LS, Green HM, Feugate JE, Yadav M, Nothnagel EA, Martins-Green M (April 2004). "Effects of "second-hand" smoke on structure and function of fibroblasts, cells that are critical for tissue repair and remodeling". BMC Cell Biology. 5 (1): 13. doi:10.1186/1471-2121-5-13. PMC 400727. PMID 15066202.
- Gosain A, DiPietro LA (March 2004). "Aging and wound healing". World Journal of Surgery. 28 (3): 321–6. doi:10.1007/s00268-003-7397-6. PMID 14961191.
- Swift ME, Burns AL, Gray KL, DiPietro LA (November 2001). "Age-related alterations in the inflammatory response to dermal injury". The Journal of Investigative Dermatology. 117 (5): 1027–35. doi:10.1046/j.0022-202x.2001.01539.x. PMID 11710909.
- Min S, Wang SW, Orr W (2006). "Graphic general pathology: 2.3 Incomplete regeneration". Pathology. pathol.med.stu.edu.cn. Archived from the original on 2013-11-10. Retrieved 2012-12-07.
The new tissue is not the same as the tissue that was lost. After the repair process has been completed, there is a loss in the structure or function of the injured tissue. In this type of repair, it is common that granulation tissue (stromal connective tissue) proliferates to fill the defect created by the necrotic cells. The necrotic cells are then replaced by scar tissue.
- Min S, Wang SW, Orr W (2006). "Graphic general pathology: 2.2 complete regeneration". Pathology. pathol.med.stu.edu.cn. Archived from the original on 2012-12-07. Retrieved 2012-12-07.
(1) Complete regeneration: The new tissue is the same as the tissue that was lost. After the repair process has been completed, the structure and function of the injured tissue are completely normal
- Min S, Wang SW, Orr W (2006). "Graphic general pathology: 2.2 complete regeneration". Pathology. pathol.med.stu.edu.cn. Archived from the original on 2012-12-07. Retrieved 2013-11-10.
After the repair process has been completed, the structure and function of the injured tissue are completely normal. This type of regeneration is common in physiological situations. Examples of physiological regeneration are the continual replacement of cells of the skin and repair of the endometrium after menstruation. Complete regeneration can occur in pathological situations in tissues that have good regenerative capacity.
- Yannas IV, Lee E, Orgill DP, Skrabut EM, Murphy GF (February 1989). "Synthesis and characterization of a model extracellular matrix that induces partial regeneration of adult mammalian skin". Proceedings of the National Academy of Sciences of the United States of America. 86 (3): 933–7. Bibcode:1989PNAS...86..933Y. doi:10.1073/pnas.86.3.933. JSTOR 33315. PMC 286593. PMID 2915988.
- O'Leary R, Ponnambalam S, Wood EJ (September 2003). "Pioglitazone-induced myofibroblast cell death: implications for cutaneous scarring". The British Journal of Dermatology. 149 (3): 665–7. doi:10.1046/j.1365-2133.2003.05501.x. PMID 14511015.
- Tong M, Tuk B, Hekking IM, Vermeij M, Barritault D, van Neck JW (2009). "Stimulated neovascularization, inflammation resolution and collagen maturation in healing rat cutaneous wounds by a heparan sulfate glycosaminoglycan mimetic, OTR4120". Wound Repair and Regeneration. 17 (6): 840–52. doi:10.1111/j.1524-475X.2009.00548.x. PMID 19903305.
- Barritault D, Caruelle JP (March 2006). "[Regenerating agents (RGTAs): a new therapeutic approach]" [Regenerating agents (RGTAs): a new therapeutic approach]. Annales Pharmaceutiques Francaises (in French). 64 (2): 135–44. doi:10.1016/S0003-4509(06)75306-8. PMID 16568015.
- Van Neck et al, Heparan sulfate proteoglycan mimetics thrive tissue regeneration: an overview. In Intech book under the working title "Tissue Regeneration", ISBN 978-953-307-876-2 is scheduled for on line publication on Nov 26, 2011"
- eurekalert.org staff (3 June 2015). "Scientist at LIMR leads study demonstrating drug-induced tissue regeneration". eurekalert.org. Lankenau Institute for Medical Research (LIMR). Archived from the original on 4 July 2015. Retrieved 3 July 2015.
- Zhang Y, Strehin I, Bedelbaeva K, Gourevitch D, Clark L, Leferovich J, Messersmith PB, Heber-Katz E. Drug-induced regeneration in adult mice. Sci Transl Med. 2015;290.
- Rush, J. (2005). Spiritual tattoo: a cultural history of tattooing, piercing, scarification, branding, and implants, Frog Ltd.
- Brown BC, McKenna SP, Siddhi K, McGrouther DA, Bayat A (September 2008). "The hidden cost of skin scars: quality of life after skin scarring". Journal of Plastic, Reconstructive & Aesthetic Surgery. 61 (9): 1049–58. doi:10.1016/j.bjps.2008.03.020. PMID 18617450.
- Bayat A, McGrouther DA, Ferguson MW (January 2003). "Skin scarring". BMJ. 326 (7380): 88–92. doi:10.1136/bmj.326.7380.88. PMC 1125033. PMID 12521975.
- Clark, R. (1996). The molecular and cellular biology of wound repair, Springer Us.
- Tonnesen MG, Feng X, Clark RA (December 2000). "Angiogenesis in wound healing". The Journal of Investigative Dermatology. Symposium Proceedings. 5 (1): 40–6. doi:10.1046/j.1087-0024.2000.00014.x. PMID 11147674.
- Ferguson MW, Whitby DJ, Shah M, Armstrong J, Siebert JW, Longaker MT (April 1996). "Scar formation: the spectral nature of fetal and adult wound repair". Plastic and Reconstructive Surgery. 97 (4): 854–60. doi:10.1097/00006534-199604000-00029. PMID 8628785.
- Brockes JP, Kumar A, Velloso CP (2001). "Regeneration as an evolutionary variable". Journal of Anatomy. 199 (Pt 1–2): 3–11. doi:10.1046/j.1469-7580.2001.19910003.x. PMC 1594962. PMID 11523827.
- "Scarless Healing". Star. Christchurch, New Zealand. 1906-07-07. pp. Page 4. Archived from the original on 2013-10-08. Retrieved 2013-07-02.
- "Scarless Healing". Marlborough Express, Volume XXXIX, Issue 160. paperspast.natlib.govt.nz. 1906-07-12. pp. Page 1. Archived from the original on 2013-10-08. Retrieved 2013-07-02.
- "A Wonderful New Surgery". Reading Eagle. 1906-07-06. pp. Page 6. Archived from the original on 2016-03-12. Retrieved 2013-07-02.
- Karin M, Clevers H (January 2016). "Reparative inflammation takes charge of tissue regeneration". Nature. 529 (7586): 307–15. Bibcode:2016Natur.529..307K. doi:10.1038/nature17039. PMC 5228603. PMID 26791721.
- Vlahopoulos SA (August 2017). "Aberrant control of NF-κB in cancer permits transcriptional and phenotypic plasticity, to curtail dependence on host tissue: molecular mode". Cancer Biology & Medicine. 14 (3): 254–270. doi:10.20892/j.issn.2095-3941.2017.0029. PMC 5570602. PMID 28884042.
- Cumming BD, McElwain DL, Upton Z (January 2010). "A mathematical model of wound healing and subsequent scarring". Journal of the Royal Society, Interface. 7 (42): 19–34. doi:10.1098/rsif.2008.0536. PMC 2839370. PMID 19324672.
- Gurtner GC, Werner S, Barrandon Y, Longaker MT (May 2008). "Wound repair and regeneration". Nature. 453 (7193): 314–21. Bibcode:2008Natur.453..314G. doi:10.1038/nature07039. PMID 18480812.
- Gurtner GC, Dauskardt RH, Wong VW, Bhatt KA, Wu K, Vial IN, Padois K, Korman JM, Longaker MT (August 2011). "Improving cutaneous scar formation by controlling the mechanical environment: large animal and phase I studies". Annals of Surgery. 254 (2): 217–25. doi:10.1097/SLA.0b013e318220b159. PMID 21606834.
- Kuhl E, Steinmann P (June 2004). "Computational modeling of healing: an application of the material force method". Biomechanics and Modeling in Mechanobiology. 2 (4): 187–203. doi:10.1007/s10237-003-0034-3. PMID 14872320.
- Rouillard AD, Holmes JW (September 2012). "Mechanical regulation of fibroblast migration and collagen remodelling in healing myocardial infarcts". The Journal of Physiology. 590 (18): 4585–602. doi:10.1113/jphysiol.2012.229484. PMC 3477759. PMID 22495588.
- Velnar T, Bailey T, Smrkolj V (2009-10-01). "The wound healing process: an overview of the cellular and molecular mechanisms". The Journal of International Medical Research. 37 (5): 1528–42. doi:10.1177/147323000903700531. PMID 19930861.
- Armitage J, Lockwood S (2011-10-01). "Skin incisions and wound closure". Surgery (Oxford). Wound Management. 29 (10): 496–501. doi:10.1016/j.mpsur.2011.06.022.
- Toon, Clare D; Lusuku, Charnelle; Ramamoorthy, Rajarajan; Davidson, Brian R; Gurusamy, Kurinchi Selvan (2015-09-03). Cochrane Wounds Group (ed.). "Early versus delayed dressing removal after primary closure of clean and clean-contaminated surgical wounds". Cochrane Database of Systematic Reviews. doi:10.1002/14651858.CD010259.pub3.
- Norman G, Dumville JC, Mohapatra DP, Owens GL, Crosbie EJ (March 2016). "Antibiotics and antiseptics for surgical wounds healing by secondary intention". The Cochrane Database of Systematic Reviews. 3: CD011712. doi:10.1002/14651858.CD011712.pub2. PMC 6599835. PMID 27021482.
- Vermeulen, Hester; Ubbink, Dirk T; Goossens, Astrid; de Vos, Rien; Legemate, Dink A; Westerbos, Stijn Joël (2004-01-26). Cochrane Wounds Group (ed.). "Dressings and topical agents for surgical wounds healing by secondary intention". Cochrane Database of Systematic Reviews. doi:10.1002/14651858.CD003554.pub2.
- Dumville, Jo C; Owens, Gemma L; Crosbie, Emma J; Peinemann, Frank; Liu, Zhenmi (2015-06-04). Cochrane Wounds Group (ed.). "Negative pressure wound therapy for treating surgical wounds healing by secondary intention". Cochrane Database of Systematic Reviews. doi:10.1002/14651858.CD011278.pub2.
- Table 3-1 in: Mitchell RS, Kumar V, Abbas AK, Nelson F (2007). Robbins Basic Pathology (8th ed.). Philadelphia: Saunders. ISBN 978-1-4160-2973-1.
- Stejskalová A, Almquist BD (July 2017). "Using biomaterials to rewire the process of wound repair". Biomaterials Science. 5 (8): 1421–1434. doi:10.1039/c7bm00295e. PMC 5576529. PMID 28692083.
- Vyas KS, Vasconez HC. Wound Healing: Biologics, Skin Substitutes, Biomembranes and Scaffolds Archived 2017-04-13 at the Wayback Machine. Healthcare. 2014; 2(3):356-400.
{kind=link}
{kind=link}
External links
| Wikimedia Commons has media related to Wound healing. |