Electron-transferring-flavoprotein dehydrogenase
Electron-transferring-flavoprotein dehydrogenase (ETF dehydrogenase or electron transfer flavoprotein-ubiquinone oxidoreductase, EC 1.5.5.1) is an enzyme that transfers electrons from electron-transferring flavoprotein in the mitochondrial matrix, to the ubiquinone pool in the inner mitochondrial membrane.[1][2] It is part of the electron transport chain. The enzyme is found in both prokaryotes and eukaryotes and contains a flavin and FE-S cluster.[3] In humans, it is encoded by the ETFDH gene. Deficiency in ETF dehydrogenase causes the human genetic disease multiple acyl-CoA dehydrogenase deficiency.[4]
| Electron-transferring-flavoprotein dehydrogenase | |
|---|---|
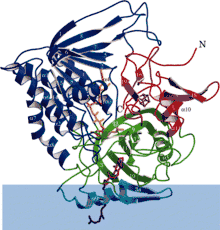 Ribbon diagram of electron-transferring-flavoprotein dehydrogenase with each functional domain differentially colored. Blue band is membrane area. | |
| Identifiers | |
| Symbol | ETFD |
| Alt. symbols | ETF-QO |
| NCBI gene | 2110 |
| HGNC | 3483 |
| OMIM | 231675 |
| PDB | 2GMH |
| RefSeq | NM_004453 |
| UniProt | Q16134 |
| Other data | |
| EC number | 1.5.5.1 |
| Locus | Chr. 4 q4q32.1 |
Function
ETQ-QO links the oxidation of fatty acids and some amino acids to oxidative phosphorylation in the mitochondria. Specifically, it catalyzes the transfer of electrons from electron transferring flavoprotein (ETF) to ubiquinone, reducing it to ubiquinol. The entire sequence of transfer reactions is as follows:[5]
Acyl-CoA → Acyl-CoA dehydrogenase → ETF → ETF-QO → UQ → Complex III.
Catalyzed reaction
The overall reaction catalyzed by ETF-QO is as follows:[6]
ETF-QO(red) + ubiquinone ↔ ETF-QO(ox) + ubiquinol
Enzymatic activity is usually assayed spectrophotometrically by reaction with octanoyl-CoA as the electron donor and ubiquinone-1 as the electron acceptor. The enzyme can also be assayed via disproportionation of ETF semiquinone. Both reactions are below:[7][8]
Octanoyl-CoA + Q1 ↔ Q1H2 + Oct-2-enoyl-CoA
2 ETF1- ↔ ETFox + ETF2-
Structure

ETF-QO consists of one structural domain with three functional domains packed in close proximity: a FAD domain, a 4Fe4S cluster domain, and a UQ-binding domain.[5] FAD is in an extended conformation and is buried deeply within its functional domain. Multiple hydrogen bonds and a positive helix dipole modulate the redox potential of FAD and can possibly stabilize the anionic semiquinone intermediate. The 4Fe4S cluster is also stabilized by extensive hydrogen bonding around the cluster and its cysteine components. Ubiquinone binding is achieved through a deep hydrophobic binding pocket which is a different mode than other UQ-binding proteins such as succinate-Q oxidoreductase. Although ETF-QO is an integral membrane protein, it does not traverse the entire membrane unlike other UQ-binding proteins.[5]
Mechanism
The exact mechanism for the reduction is unknown, although there are two hypothesized pathways. The first pathway is the transferral of electrons from one electron reduced ETF one at a time to the lower potential FAD center. One electron is transferred from the reduced FAD to the iron cluster, resulting in a two electron reduced state with one electron each on the FAD and cluster domains. Then, the bound ubiquinone is reduced to ubiquinol, at least transiently forming the singly reduced semiubiquinone. The second pathway involves the donation of electrons from ETF to the iron cluster, followed by internal transitions between the two electron centers. After equilibration, the rest of the pathway follows as above.[5]
Clinical significance
Deficiency of ETF-QO results in a disorder known as glutaric acidemia type II (also known as MADD for multiple acyl-CoA dehydrogenase deficiency), in which there is an improper buildup of fats and proteins in the body.[9] Complications can involve acidosis or hypoglycemia, with other symptoms such as general weakness, liver enlargement, increased heart failure, and carnitine deficiency. More severe cases involve congenital defects and full metabolic crisis.[10][11][12] Genetically, it is an autosomal recessive disorder, making its occurrence fairly rare. Most affected patients are the result of single point mutations around the FAD ubiquinone interface.[13][14] Milder forms of the disorder have been responsive to riboflavin therapy and are coined riboflavin-responsive MADD (RR-MADD), although due to the varying mutations causing the disease treatment and symptoms can vary considerably.[15][16]
References
- Ghisla S, Thorpe C (Feb 2004). "Acyl-CoA dehydrogenases. A mechanistic overview". European Journal of Biochemistry / FEBS. 271 (3): 494–508. doi:10.1046/j.1432-1033.2003.03946.x. PMID 14728676.
- He M, Rutledge SL, Kelly DR, Palmer CA, Murdoch G, Majumder N, Nicholls RD, Pei Z, Watkins PA, Vockley J (Jul 2007). "A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency". American Journal of Human Genetics. 81 (1): 87–103. doi:10.1086/519219. PMC 1950923. PMID 17564966.
- Watmough NJ, Frerman FE (Dec 2010). "The electron transfer flavoprotein: ubiquinone oxidoreductases". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1797 (12): 1910–6. doi:10.1016/j.bbabio.2010.10.007. PMID 20937244.
- Vianey-Liaud C, Divry P, Gregersen N, Mathieu M (1987). "The inborn errors of mitochondrial fatty acid oxidation". Journal of Inherited Metabolic Disease. 10 Suppl 1: 159–200. doi:10.1007/bf01812855. PMID 3119938.
- Zhang J, Frerman FE, Kim JJ (Oct 2006). "Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool". Proceedings of the National Academy of Sciences of the United States of America. 103 (44): 16212–7. doi:10.1073/pnas.0604567103. PMC 1637562. PMID 17050691.
- Ramsay RR, Steenkamp DJ, Husain M (Feb 1987). "Reactions of electron-transfer flavoprotein and electron-transfer flavoprotein: ubiquinone oxidoreductase". The Biochemical Journal. 241 (3): 883–92. doi:10.1042/bj2410883. PMC 1147643. PMID 3593226.
- Beckmann JD, Frerman FE (Jul 1985). "Reaction of electron-transfer flavoprotein with electron-transfer flavoprotein-ubiquinone oxidoreductase". Biochemistry. 24 (15): 3922–5. doi:10.1021/bi00336a017. PMID 2996585.
- Watmough NJ, Loehr JP, Drake SK, Frerman FE (Feb 1991). "Tryptophan fluorescence in electron-transfer flavoprotein:ubiquinone oxidoreductase: fluorescence quenching by a brominated pseudosubstrate". Biochemistry. 30 (5): 1317–23. doi:10.1021/bi00219a023. PMID 1991113.
- Frerman, F. E.; Goodman, S. I. (1985). "Deficiency of Electron Transfer Flavoprotein or Electron Transfer Flavoprotein:Ubiquinone Oxidoreductase in Glutaric Acidemia Type II Fibroblasts". Proceedings of the National Academy of Sciences. 82 (13): 4517–4520. doi:10.1073/pnas.82.13.4517. PMC 391133. PMID 2989828.
- Galloway JH, Cartwright IJ, Bennett MJ (Mar 1987). "Abnormal myocardial lipid composition in an infant with type II glutaric aciduria". Journal of Lipid Research. 28 (3): 279–84. PMID 3572253.
- Singla M, Guzman G, Griffin AJ, Bharati S (Mar 2008). "Cardiomyopathy in multiple Acyl-CoA dehydrogenase deficiency: a clinico-pathological correlation and review of literature". Pediatric Cardiology. 29 (2): 446–51. doi:10.1007/s00246-007-9119-6. PMID 17912479.
- Turnbull DM, Bartlett K, Eyre JA, Gardner-Medwin D, Johnson MA, Fisher J, Watmough NJ (Oct 1988). "Lipid storage myopathy due to glutaric aciduria type II: treatment of a potentially fatal myopathy". Developmental Medicine and Child Neurology. 30 (5): 667–72. doi:10.1111/j.1469-8749.1988.tb04806.x. PMID 3229565.
- Liang WC, Ohkuma A, Hayashi YK, López LC, Hirano M, Nonaka I, Noguchi S, Chen LH, Jong YJ, Nishino I (Mar 2009). "ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency". Neuromuscular Disorders. 19 (3): 212–6. doi:10.1016/j.nmd.2009.01.008. PMID 19249206.
- Goodman SI, Binard RJ, Woontner MR, Frerman FE (2002). "Glutaric acidemia type II: gene structure and mutations of the electron transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO) gene". Molecular Genetics and Metabolism. 77 (1–2): 86–90. doi:10.1016/S1096-7192(02)00138-5. PMID 12359134.
- Olsen RK, Olpin SE, Andresen BS, Miedzybrodzka ZH, Pourfarzam M, Merinero B, Frerman FE, Beresford MW, Dean JC, Cornelius N, Andersen O, Oldfors A, Holme E, Gregersen N, Turnbull DM, Morris AA (Aug 2007). "ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency". Brain. 130 (Pt 8): 2045–54. doi:10.1093/brain/awm135. PMID 17584774.
- Rhead W, Roettger V, Marshall T, Amendt B (Feb 1993). "Multiple acyl-coenzyme A dehydrogenation disorder responsive to riboflavin: substrate oxidation, flavin metabolism, and flavoenzyme activities in fibroblasts". Pediatric Research. 33 (2): 129–35. doi:10.1203/00006450-199302000-00008. PMID 8433888.