Enzyme catalysis
Enzyme catalysis is the increase in the rate of a process by a biological molecule, an "enzyme". Most enzymes are proteins, and most such processes are chemical reactions. Within the enzyme, generally catalysis occurs at a localized site, called the active site.
Most enzymes are made predominantly of proteins, either a single protein chain or many such chains in a multi-subunit complex. Enzymes often also incorporate non-protein components, such as metal ions or specialized organic molecules known as cofactor (e.g. adenosine triphosphate). Many cofactors are vitamins, and their role as vitamins is directly linked to their use in the catalysis of biological process within metabolism. Catalysis of biochemical reactions in the cell is vital since many but not all metabolically essential reactions have very low rates when uncatalysed. One driver of protein evolution is the optimization of such catalytic activities, although only the most crucial enzymes operate near catalytic efficiency limits, and many enzymes are far from optimal. Important factors in enzyme catalysis include general acid and base catalysis, orbital steering, entropic restriction, orientation effects (i.e. lock and key catalysis), as well as motional effects involving protein dynamics[1]
Mechanisms of enzyme catalysis vary, but are all similar in principle to other types of chemical catalysis in that the crucial factor is a reduction of energy barrier(s) separating the reactants from the products. The reduction of activation energy (Ea) increases the fraction of reactant molecules that can overcome this barrier and form the product. An important principle is that since they only reduce energy barriers between products and reactants, enzymes always catalyze reactions in both directions, and cannot drive a reaction forward or affect the equilibrium position - only the speed with which is it achieved. As with other catalysts, the enzyme is not consumed or changed by the reaction (as a substrate is) but is recycled such that a single enzyme performs many rounds of catalysis.
Induced fit

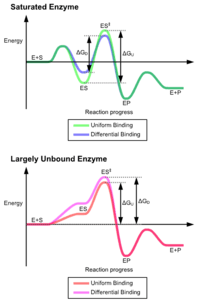
The classic model for the enzyme-substrate interaction is the induced fit model.[2] This model proposes that the initial interaction between enzyme and substrate is relatively weak, but that these weak interactions rapidly induce conformational changes in the enzyme that strengthen binding.
The advantages of the induced fit mechanism arise due to the stabilizing effect of strong enzyme binding. There are two different mechanisms of substrate binding: uniform binding, which has strong substrate binding, and differential binding, which has strong transition state binding. The stabilizing effect of uniform binding increases both substrate and transition state binding affinity, while differential binding increases only transition state binding affinity. Both are used by enzymes and have been evolutionarily chosen to minimize the activation energy of the reaction. Enzymes that are saturated, that is, have a high affinity substrate binding, require differential binding to reduce the energy of activation, whereas small substrate unbound enzymes may use either differential or uniform binding.[3]
These effects have led to most proteins using the differential binding mechanism to reduce the energy of activation, so most substrates have high affinity for the enzyme while in the transition state. Differential binding is carried out by the induced fit mechanism - the substrate first binds weakly, then the enzyme changes conformation increasing the affinity to the transition state and stabilizing it, so reducing the activation energy to reach it.
It is important to clarify, however, that the induced fit concept cannot be used to rationalize catalysis. That is, the chemical catalysis is defined as the reduction of Ea‡ (when the system is already in the ES‡) relative to Ea‡ in the uncatalyzed reaction in water (without the enzyme). The induced fit only suggests that the barrier is lower in the closed form of the enzyme but does not tell us what the reason for the barrier reduction is.
Induced fit may be beneficial to the fidelity of molecular recognition in the presence of competition and noise via the conformational proofreading mechanism .[4]
Mechanisms of an alternative reaction route
These conformational changes also bring catalytic residues in the active site close to the chemical bonds in the substrate that will be altered in the reaction. After binding takes place, one or more mechanisms of catalysis lowers the energy of the reaction's transition state, by providing an alternative chemical pathway for the reaction. There are six possible mechanisms of "over the barrier" catalysis as well as a "through the barrier" mechanism:
Proximity and orientation
Enzyme-substrate interactions align the reactive chemical groups and hold them close together in an optimal geometry, which increases the rate of the reaction. This reduces the entropy of the reactants and thus makes addition or transfer reactions less unfavorable, since a reduction in the overall entropy when two reactants become a single product. However this is a general effect and is seen in non-addition or transfer reactions where it occurs due to an increase in the "effective concentration" of the reagents. This is understood when considering how increases in concentration leads to increases in reaction rate: essentially when the reactants are more concentrated, they collide more often and so react more often. In enzyme catalysis, the binding of the reagents to the enzyme restricts the conformational space of the reactants, holding them in the 'proper orientation' and close to each other, so that the collide more frequently, and with the correct geometry, to facilitate the desired reaction. The "effective concentration" is the concentration the reactant would have to be, free in solution, to experiences the same collisional frequency. Often such theoretical effective concentrations are unphysical and impossible to realize in reality - which is a testament to the great catalytic power of many enzymes, with massive rate increases over the uncatalyzed state.
| For example: |
| Similar reactions will occur far faster if the reaction is intramolecular. |
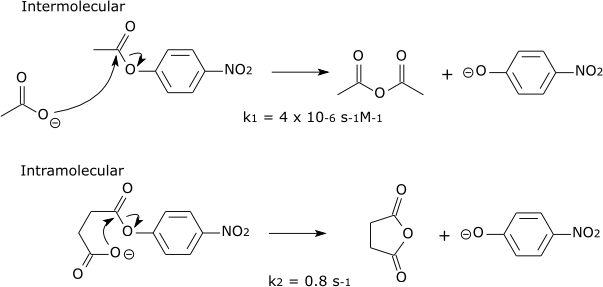 |
| The effective concentration of acetate in the intramolecular reaction can be estimated as k2/k1 = 2 x 105 Molar. |
However, the situation might be more complex, since modern computational studies have established that traditional examples of proximity effects cannot be related directly to enzyme entropic effects.[5][6][7] Also, the original entropic proposal[8] has been found to largely overestimate the contribution of orientation entropy to catalysis.[9]
Proton donors or acceptors
Proton donors and acceptors, i.e. acids and base may donate and accept protons in order to stabilize developing charges in the transition state. This is related to the overall principle of catalysis, that of reducing energy barriers, since in general transition stated are high energy states, and by stabilizing them this high energy is reduced, lowering the barrier. A key feature of enzyme catalysis over many non-biological catalysis, is that both acid and base catalysis can be combined in the same reaction. In many abiotic systems, acids (large [H+]) or bases ( large concentration H+ sinks, or species with electron pairs) can increase the rate of the reaction; but of course the environment can only have one overall pH (measure of acidity or basicity (alkalinity)). However since enzymes are large molecules, they can position both acid groups and basic groups in their active site to interact with their substrates, and employ both modes independent of the bulk pH.
Often general acid or base catalysis is employed to activate nucleophile and/or electrophile groups, or to stabilizing leaving groups. Many amino acids with acidic or basic groups are this employed in the active site, such as the glutamic and aspartic acid, histidine, cystine, tyrosine, lysine and arginine, as well as serine and threonine. In addition, the peptide backbone, with carbonyl and amide N groups is often employed. Cystine and Histidine are very commonly involved, since they both have a pKa close to neutral pH and can therefore both accept and donate protons.
Many reaction mechanisms involving acid/base catalysis assume a substantially altered pKa. This alteration of pKa is possible through the local environment of the residue.
| Conditions | Acids | Bases |
|---|---|---|
| Hydrophobic environment | Increase pKa | Decrease pKa |
| Adjacent residues of like charge | Increase pKa | Decrease pKa |
| Salt bridge (and hydrogen bond) formation |
Decrease pKa | Increase pKa |
pKa can also be influenced significantly by the surrounding environment, to the extent that residues which are basic in solution may act as proton donors, and vice versa.
| For example: |
| Catalytic triad of a serine protease |
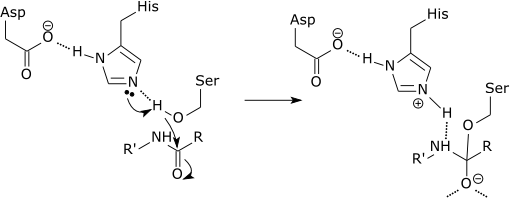 |
| The initial step of the serine protease catalytic mechanism involves the histidine of the active site accepting a proton from the serine residue. This prepares the serine as a nucleophile to attack the amide bond of the substrate. This mechanism includes donation of a proton from serine (a base, pKa 14) to histidine (an acid, pKa 6), made possible due to the local environment of the bases. |
It is important to clarify that the modification of the pKa's is a pure part of the electrostatic mechanism.[10] Furthermore, the catalytic effect of the above example is mainly associated with the reduction of the pKa of the oxyanion and the increase in the pKa of the histidine, while the proton transfer from the serine to the histidine is not catalyzed significantly, since it is not the rate determining barrier.[11]
Electrostatic catalysis
Stabilization of charged transition states can also be by residues in the active site forming ionic bonds (or partial ionic charge interactions) with the intermediate. These bonds can either come from acidic or basic side chains found on amino acids such as lysine, arginine, aspartic acid or glutamic acid or come from metal cofactors such as zinc. Metal ions are particularly effective and can reduce the pKa of water enough to make it an effective nucleophile.
Systematic computer simulation studies established that electrostatic effects give, by far, the largest contribution to catalysis.[10] It can increase the rate of reaction by a factor of up to 107.[12] In particular, it has been found that enzyme provides an environment which is more polar than water, and that the ionic transition states are stabilized by fixed dipoles. This is very different from transition state stabilization in water, where the water molecules must pay with "reorganization energy".[13] In order to stabilize ionic and charged states. Thus, the catalysis is associated with the fact that the enzyme polar groups are preorganized [14]
The magnitude of the electrostatic field exerted by an enzyme's active site has been shown to be highly correlated with the enzyme's catalytic rate enhancement[15][16]
Binding of substrate usually excludes water from the active site, thereby lowering the local dielectric constant to that of an organic solvent. This strengthens the electrostatic interactions between the charged/polar substrates and the active sites. In addition, studies have shown that the charge distributions about the active sites are arranged so as to stabilize the transition states of the catalyzed reactions. In several enzymes, these charge distributions apparently serve to guide polar substrates toward their binding sites so that the rates of these enzymatic reactions are greater than their apparent diffusion-controlled limits.
| For example: |
| Carboxypeptidase catalytic mechanism |
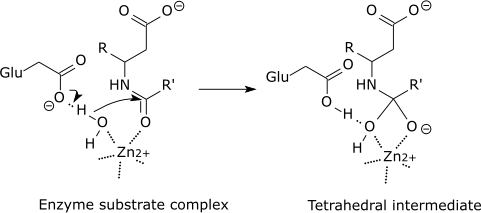 |
| The tetrahedral intermediate is stabilised by a partial ionic bond between the Zn2+ ion and the negative charge on the oxygen. |
Covalent catalysis
Covalent catalysis involves the substrate forming a transient covalent bond with residues in the enzyme active site or with a cofactor. This adds an additional covalent intermediate to the reaction, and helps to reduce the energy of later transition states of the reaction. The covalent bond must, at a later stage in the reaction, be broken to regenerate the enzyme. This mechanism is utilised by the catalytic triad of enzymes such as proteases like chymotrypsin and trypsin, where an acyl-enzyme intermediate is formed. An alternative mechanism is schiff base formation using the free amine from a lysine residue, as seen in the enzyme aldolase during glycolysis.
Some enzymes utilize non-amino acid cofactors such as pyridoxal phosphate (PLP) or thiamine pyrophosphate (TPP) to form covalent intermediates with reactant molecules.[17][18] Such covalent intermediates function to reduce the energy of later transition states, similar to how covalent intermediates formed with active site amino acid residues allow stabilization, but the capabilities of cofactors allow enzymes to carryout reactions that amino acid side residues alone could not. Enzymes utilizing such cofactors include the PLP-dependent enzyme aspartate transaminase and the TPP-dependent enzyme pyruvate dehydrogenase.[19][20]
Rather than lowering the activation energy for a reaction pathway, covalent catalysis provides an alternative pathway for the reaction (via to the covalent intermediate) and so is distinct from true catalysis.[10] For example, the energetics of the covalent bond to the serine molecule in chymotrypsin should be compared to the well-understood covalent bond to the nucleophile in the uncatalyzed solution reaction. A true proposal of a covalent catalysis (where the barrier is lower than the corresponding barrier in solution) would require, for example, a partial covalent bond to the transition state by an enzyme group (e.g., a very strong hydrogen bond), and such effects do not contribute significantly to catalysis.
Metal ion catalysis
A metal ion in the active site participates in catalysis by coordinating charge stabilization and shielding. Because of a metal's positive charge, only negative charges can be stabilized through metal ions.[21] However, metal ions are advantageous in biological catalysis because they are not affected by changes in pH.[22] Metal ions can also act to ionize water by acting as a Lewis acid.[23] Metal ions may also be agents of oxidation and reduction.[24]
Bond strain
This is the principal effect of induced fit binding, where the affinity of the enzyme to the transition state is greater than to the substrate itself. This induces structural rearrangements which strain substrate bonds into a position closer to the conformation of the transition state, so lowering the energy difference between the substrate and transition state and helping catalyze the reaction.
However, the strain effect is, in fact, a ground state destabilization effect, rather than transition state stabilization effect.[10][25] Furthermore, enzymes are very flexible and they cannot apply large strain effect.[26]
In addition to bond strain in the substrate, bond strain may also be induced within the enzyme itself to activate residues in the active site.
| For example: |
| Substrate, bound substrate, and transition state conformations of lysozyme. |
| The substrate, on binding, is distorted from the half chair conformation of the hexose ring (because of the steric hindrance with amino acids of the protein forcing the equatorial c6 to be in the axial position) into the chair conformation[27] |
Quantum tunneling
These traditional "over the barrier" mechanisms have been challenged in some cases by models and observations of "through the barrier" mechanisms (quantum tunneling). Some enzymes operate with kinetics which are faster than what would be predicted by the classical ΔG‡. In "through the barrier" models, a proton or an electron can tunnel through activation barriers.[28][29] Quantum tunneling for protons has been observed in tryptamine oxidation by aromatic amine dehydrogenase.[30]
Quantum tunneling does not appear to provide a major catalytic advantage, since the tunneling contributions are similar in the catalyzed and the uncatalyzed reactions in solution.[29][31][32][33] However, the tunneling contribution (typically enhancing rate constants by a factor of ~1000[30] compared to the rate of reaction for the classical 'over the barrier' route) is likely crucial to the viability of biological organisms. This emphasizes the general importance of tunneling reactions in biology.
In 1971-1972 the first quantum-mechanical model of enzyme catalysis was formulated.[34][35]
Active enzyme
The binding energy of the enzyme-substrate complex cannot be considered as an external energy which is necessary for the substrate activation. The enzyme of high energy content may firstly transfer some specific energetic group X1 from catalytic site of the enzyme to the final place of the first bound reactant, then another group X2 from the second bound reactant (or from the second group of the single reactant) must be transferred to active site to finish substrate conversion to product and enzyme regeneration.[36]
We can present the whole enzymatic reaction as a two coupling reactions:
-
(1)

-
(2)

It may be seen from reaction (1) that the group X1 of the active enzyme appears in the product due to possibility of the exchange reaction inside enzyme to avoid both electrostatic inhibition and repulsion of atoms. So we represent the active enzyme as a powerful reactant of the enzymatic reaction. The reaction (2) shows incomplete conversion of the substrate because its group X2 remains inside enzyme. This approach as idea had formerly proposed relying on the hypothetical extremely high enzymatic conversions (catalytically perfect enzyme).[37]
The crucial point for the verification of the present approach is that the catalyst must be a complex of the enzyme with the transfer group of the reaction. This chemical aspect is supported by the well-studied mechanisms of the several enzymatic reactions. Consider the reaction of peptide bond hydrolysis catalyzed by a pure protein α-chymotrypsin (an enzyme acting without a cofactor), which is a well-studied member of the serine proteases family, see.[38]
We present the experimental results for this reaction as two chemical steps:
-
(3)

-
(4)

where S1 is a polypeptide, P1 and P2 are products. The first chemical step (3) includes the formation of a covalent acyl-enzyme intermediate. The second step (4) is the deacylation step. It is important to note that the group H+, initially found on the enzyme, but not in water, appears in the product before the step of hydrolysis, therefore it may be considered as an additional group of the enzymatic reaction.
Thus, the reaction (3) shows that the enzyme acts as a powerful reactant of the reaction. According to the proposed concept, the H transport from the enzyme promotes the first reactant conversion, breakdown of the first initial chemical bond (between groups P1 and P2). The step of hydrolysis leads to a breakdown of the second chemical bond and regeneration of the enzyme.
The proposed chemical mechanism does not depend on the concentration of the substrates or products in the medium. However, a shift in their concentration mainly causes free energy changes in the first and final steps of the reactions (1) and (2) due to the changes in the free energy content of every molecule, whether S or P, in water solution. This approach is in accordance with the following mechanism of muscle contraction. The final step of ATP hydrolysis in skeletal muscle is the product release caused by the association of myosin heads with actin.[39] The closing of the actin-binding cleft during the association reaction is structurally coupled with the opening of the nucleotide-binding pocket on the myosin active site.[40]
Notably, the final steps of ATP hydrolysis include the fast release of phosphate and the slow release of ADP.[41][42] The release of a phosphate anion from bound ADP anion into water solution may be considered as an exergonic reaction because the phosphate anion has low molecular mass.
Thus, we arrive at the conclusion that the primary release of the inorganic phosphate H2PO4− leads to transformation of a significant part of the free energy of ATP hydrolysis into the kinetic energy of the solvated phosphate, producing active streaming. This assumption of a local mechano-chemical transduction is in accord with Tirosh's mechanism of muscle contraction, where the muscle force derives from an integrated action of active streaming created by ATP hydrolysis.[43][44]
Examples of catalytic mechanisms
In reality, most enzyme mechanisms involve a combination of several different types of catalysis.
Triose phosphate isomerase
Triose phosphate isomerase (EC 5.3.1.1) catalyses the reversible interconvertion of the two triose phosphates isomers dihydroxyacetone phosphate and D-glyceraldehyde 3-phosphate.
Trypsin
Trypsin (EC 3.4.21.4) is a serine protease that cleaves protein substrates after lysine or arginine residues using a catalytic triad to perform covalent catalysis, and an oxyanion hole to stabilise charge-buildup on the transition states.
Aldolase
Aldolase (EC 4.1.2.13) catalyses the breakdown of fructose 1,6-bisphosphate (F-1,6-BP) into glyceraldehyde 3-phosphate and dihydroxyacetone phosphate (DHAP).
Enzyme diffusivity
The advent of single-molecule studies led in the 2010s to the observation that the movement of untethered enzymes increases with increasing substrate concentration and increasing reaction enthalpy.[45] Subsequent observations suggest that this increase in diffusivity is driven by transient displacement of the enzyme's center of mass, resulting in a "recoil effect that propels the enzyme".[46]
Reaction similarity
Similarity between enzymatic reactions (EC) can be calculated by using bond changes, reaction centres or substructure metrics (EC-BLAST).[47]
See also
- Catalytic triad
- Enzyme assay
- Enzyme kinetics
- Enzyme promiscuity
- Protein dynamics
- Pseudoenzymes, whose ubiquity despite their catalytic inactivity suggests omic implications
- Quantum tunnelling
- The Proteolysis Map
- Time resolved crystallography
References
- Kamerlin, S. C.; Warshel, A (2010). "At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis?". Proteins: Structure, Function, and Bioinformatics. 78 (6): 1339–75. doi:10.1002/prot.22654. PMC 2841229. PMID 20099310.
- Koshland DE (February 1958). "Application of a Theory of Enzyme Specificity to Protein Synthesis". Proc. Natl. Acad. Sci. U.S.A. 44 (2): 98–104. Bibcode:1958PNAS...44...98K. doi:10.1073/pnas.44.2.98. PMC 335371. PMID 16590179.

- Anslyn, E.V.; Dougherty, D.A. (2006). Modern Physical Organic Chemistry. University Science Books. ISBN 978-1-891389-31-3.
- Savir Y; Tlusty T (2007). Scalas, Enrico (ed.). "Conformational Proofreading: The Impact of Conformational Changes on the Specificity of Molecular Recognition" (PDF). PLOS One. 2 (5): e468. Bibcode:2007PLoSO...2..468S. doi:10.1371/journal.pone.0000468. PMC 1868595. PMID 17520027. Archived from the original (PDF) on 14 May 2011. Retrieved 22 August 2010.
- Stanton, R.V.; Perakyla, M.; Bakowies, D.; Kollman, P.A. (1998). "Combined ab initio and Free Energy Calculations To Study Reactions in Enzymes and Solution: Amide Hydrolysis in Trypsin and Aqueous Solution". J. Am. Chem. Soc. 120 (14): 3448–3457. doi:10.1021/ja972723x.
- Kuhn, B.; Kollman, P.A. (2000). "QM-FE and Molecular Dynamics Calculations on Catechol O-Methyltransferase: Free Energy of Activation in the Enzyme and in Aqueous Solution and Regioselectivity of the Enzyme-Catalyzed Reaction". J. Am. Chem. Soc. 122 (11): 2586–2596. doi:10.1021/ja992218v.
- Bruice, T.C.; Lightstone, F.C. (1999). "Ground State and Transition State Contributions to the Rates of Intramolecular and Enzymatic Reactions". Acc. Chem. Res. 32 (2): 127–136. doi:10.1021/ar960131y.
- Page, M.I.; Jencks, W.P. (1971). "Entropic Contributions to Rate Accelerations in Enzymic and Intramolecular Reactions and the Chelate Effect". Proc. Natl. Acad. Sci. USA. 68 (8): 1678–1683. Bibcode:1971PNAS...68.1678P. doi:10.1073/pnas.68.8.1678. PMC 389269. PMID 5288752.
- Warshel, A.; Parson, W.W. (2001). "Dynamics of Biochemical and Biophysical Reactions: Insight from Computer Simulations". Quart. Rev. Biophys. 34 (4): 563–679. doi:10.1017/s0033583501003730.
- Warshel, A.; Sharma, P.K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M.H.M. (2006). "Electrostatic Basis of Enzyme Catalysis". Chem. Rev. 106 (8): 3210–3235. doi:10.1021/cr0503106. PMID 16895325.
- Warshel, A.; Naray-Szabo, G.; Sussman, F.; Hwang, J.-K. (1989). "How do Serine Proteases Really Work?". Biochemistry. 28 (9): 3629–37. doi:10.1021/bi00435a001. PMID 2665806.
- Voet, Donald. (cop. 2011). Biochemistry. John Wiley & Sons. OCLC 808679090. Check date values in:
|date=(help) - Marcus, R. A. (1965). "On the Theory of Electron-Transfer Reactions. VI. Unified Treatment for Homogeneous and Electrode Reactions" (PDF). J. Chem. Phys. 43 (2): 679–701. Bibcode:1965JChPh..43..679M. doi:10.1063/1.1696792.
- Warshel, A (1978). "Energetics of Enzyme Catalysis". Proc. Natl. Acad. Sci. USA. 75 (11): 5250–5254. Bibcode:1978PNAS...75.5250W. doi:10.1073/pnas.75.11.5250. PMC 392938. PMID 281676.
- "How Enzymes Work"
- "EXTREME ELECTRIC FIELDS POWER CATALYSIS IN THE ACTIVE SITE OF KETOSTEROID ISOMERASE",
- Toney, M. D. "Reaction specificity in pyridoxal enzymes." Archives of biochemistry and biophysics (2005) 433: 279-287
- Micronutrient Information Center, Oregon State University
- Voet, Donald; Judith Voet (2004). Biochemistry. John Wiley & Sons Inc. pp. 986–989. ISBN 978-0-471-25090-6.
- Voet, Donald; Judith Voet (2004). Biochemistry. John Wiley & Sons Inc. pp. 604–606. ISBN 978-0-471-25090-6.
- Piccirilli, Joseph A.; Vyle, Joseph S.; Caruthers, Marvin H.; Cech, Thomas R. (7 January 1993). "Metal ion catalysis in the Tetrahymena ribozyme reaction". Nature. 361 (6407): 85–88. Bibcode:1993Natur.361...85P. doi:10.1038/361085a0. PMID 8421499.
- BENDER, MYRON L. (1 January 1962). "Metal Ion Catalysis of Nucleophilic Organic Reactions in Solution". Reactions of Coordinated Ligands. Advances in Chemistry. 37. AMERICAN CHEMICAL SOCIETY. pp. 19–36. doi:10.1021/ba-1963-0037.ch002. ISBN 978-0841200388.
- Fife, Thomas H.; Przystas, Theodore J. (1 February 1985). "Divalent metal ion catalysis in the hydrolysis of esters of picolinic acid. Metal ion promoted hydroxide ion and water catalyzed reactions". Journal of the American Chemical Society. 107 (4): 1041–1047. doi:10.1021/ja00290a048. ISSN 0002-7863.
- Stadtman, E. R. (1 January 1990). "Metal ion-catalyzed oxidation of proteins: biochemical mechanism and biological consequences". Free Radical Biology & Medicine. 9 (4): 315–325. doi:10.1016/0891-5849(90)90006-5. ISSN 0891-5849. PMID 2283087.
- Jencks, William P. (1987) [1969]. Catalysis in Chemistry and Enzymology. McGraw-Hill series in advanced chemistry (reprint ed.). New York: Dover Publications. ISBN 9780486654607.
- Warshel, A.; Levitt, M. (1976). "Theoretical Studies of Enzymatic Reactions: Dielectric Electrostatic and Steric Stabilization of the Carbonium Ion in the Reaction of Lysozyme". Journal of Molecular Biology. 103 (2): 227–49. doi:10.1016/0022-2836(76)90311-9. PMID 985660.
- <fundamentals of biochemistry Voet, voet and Pratt 4th edition>, which is similar in shape to the transition state.
- Garcia-Viloca, M; Gao, J; Karplus, M; Truhlar, DG (2004). "How enzymes work: analysis by modern rate theory and computer simulations". Science. 303 (5655): 186–95. Bibcode:2004Sci...303..186G. doi:10.1126/science.1088172. PMID 14716003.
- Olsson, MH; Siegbahn, PE; Warshel, A (2004). "Simulations of the large kinetic isotope effect and the temperature dependence of the hydrogen atom transfer in lipoxygenase". Journal of the American Chemical Society. 126 (9): 2820–8. doi:10.1021/ja037233l. PMID 14995199.
- Masgrau, L; Roujeinikova, A; Johannissen, LO; Hothi, P; Basran, J; Ranaghan, KE; Mulholland, AJ; Sutcliffe, MJ; et al. (2006). "Atomic description of an enzyme reaction dominated by proton tunneling". Science. 312 (5771): 237–41. Bibcode:2006Sci...312..237M. doi:10.1126/science.1126002. PMID 16614214.
- Hwang, J.-K.; Warshel, A. (1996). "How important are quantum mechanical nuclear motions in enzyme catalysis". J. Am. Chem. Soc. 118 (47): 11745–11751. doi:10.1021/ja962007f.
- Ball, P. (2004). "Enzymes: By chance, or by design?". Nature. 431 (7007): 396–397. Bibcode:2004Natur.431..396B. doi:10.1038/431396a. PMID 15385982.
- Olsson, M.H.M.; Parson, W.W.; Warshel, A. (2006). "Dynamical Contributions to Enzyme Catalysis: Critical Tests of A Popular Hypothesis". Chem. Rev. 106 (5): 1737–1756. doi:10.1021/cr040427e. PMID 16683752.
- Volkenshtein M.V., Dogonadze R.R., Madumarov A.K., Urushadze Z.D., Kharkats Yu.I. Theory of Enzyme Catalysis.- Molekuliarnaya Biologia, Moscow, 6, 1972, 431-439
- Volkenshtein M.V., Dogonadze R.R., Madumarov A.K., Urushadze Z.D., Kharkats Yu.I. Electronic and Conformational Interactions in Enzyme Catalysis. In: E.L. Andronikashvili (Ed.), Konformatsionnie Izmenenia Biopolimerov v Rastvorakh, Publishing House "Nauka", Moscow, 1973, 153-157
- Foigel, Alexander G. (2011). "Is the enzyme a powerful reactant of the biochemical reaction?". Molecular and Cellular Biochemistry. 352 (1–2): 87–9. doi:10.1007/s11010-011-0742-4. PMID 21318350.
- Fogel, A.G. (1982). "Cooperativity of enzymatic reactions and molecular aspects of energy transduction". Mol. Cell. Biochem. 47 (1): 59–64. doi:10.1007/bf00241567. PMID 7132966.
- Hengge, AC; Stein, RL (2004). "Role of protein conformational mobility in enzyme catalysis: acylation of alpha-chymotrypsin by specific peptide substrates". Biochemistry. 43 (3): 742–747. doi:10.1021/bi030222k. PMID 14730979.
- Lymn, RW; Taylor, EW. (1971). "Mechanism of adenosine triphosphate hydrolysis by actomyosin". Biochemistry. 10 (25): 4617–4624. doi:10.1021/bi00801a004. PMID 4258719.
- Holmes, KC; Angert, I; Kull, FG; Jahn, W; Schroder, RR. (2003). "Electron cryo-microscopy shows how strong binding of myosin to actin releases nucleotide". Nature. 425 (6956): 423–427. Bibcode:2003Natur.425..423H. doi:10.1038/nature02005. PMID 14508495.
- Siemankowski, RF; Wiseman, MO; White, HD. (1985). "ADP dissociation from actomyosin subfragment 1 is sufficiently slow to limit the unloaded shortening velocity in vertebrate muscle". Proc. Natl. Acad. Sci. USA. 82 (3): 658–662. Bibcode:1985PNAS...82..658S. doi:10.1073/pnas.82.3.658. PMC 397104. PMID 3871943.
- White, HD; Belknap, B; Webb, MR. (1997). "Kinetics of nucleoside triphosphate cleavage and phosphate release steps by associated rabbit skeletal actomyosin, measured using a novel fluorescent probe for phosphate". Biochemistry. 36 (39): 11828–11836. doi:10.1021/bi970540h. PMID 9305974.
- Tirosh, R; Low, WZ; Oplatka, A. (1990). "Translational motion of actin filaments in the presence of heavy meromyosin and MgATP as measured by Doppler broadening of laser light scattering". Biochim. Biophys. Acta. 1037 (3): 274–280. doi:10.1016/0167-4838(90)90025-b. PMID 2178685.
- Tirosh, R. (2006). "Ballistic protons and microwave-induced water solutions (solitons) in bioenergetic transformations". Int. J. Mol. Sci. 7 (9): 320–345. doi:10.3390/i7090320.
- Muddana, Hari S.; Sengupta, Samudra; Mallouk, Thomas E.; et al. (28 January 2010). "Substrate Catalysis Enhances Single-Enzyme Diffusion". Journal of the American Chemical Society. 132 (7): 2110–1. doi:10.1021/ja908773a. PMC 2832858. PMID 20108965.

- Riedel, Clement; Gabizon, Ronen; Wilson, Christian A. M.; et al. (8 January 2015). "The heat released during catalytic turnover enhances the diffusion of an enzyme". Nature. 517 (7533): 227–30. Bibcode:2015Natur.517..227R. doi:10.1038/nature14043. PMC 4363105. PMID 25487146. Lay summary – Nature: News & Views (8 January 2015).
- Rahman, SA; Cuesta, SM; Furnham, N; Holliday, GL; Thornton, JM (2014). "EC-BLAST: a tool to automatically search and compare enzyme reactions". Nature Methods. 11 (2): 171–174. doi:10.1038/nmeth.2803. PMC 4122987. PMID 24412978.
Further reading
- Alan Fersht, Structure and Mechanism in Protein Science : A Guide to Enzyme Catalysis and Protein Folding. W. H. Freeman, 1998. ISBN 0-7167-3268-8
- Dedicated issue of Philosophical Transactions B on Quantum catalysis in enzymes freely available.
External links
