TIM barrel
The TIM barrel is a conserved protein fold consisting of eight α-helices and eight parallel β-strands that alternate along the peptide backbone.[1] The structure is named after triosephosphate isomerase, a conserved metabolic enzyme. TIM barrels are ubiquitous, with approximately 10% of all enzymes adopting this fold.[3] Further, 5 of 7 enzyme commission (EC) enzyme classes include TIM barrel proteins.[4][5] The TIM barrel fold is evolutionarily ancient, with many of its members possessing little similarity today,[6] instead falling within the twilight zone of sequence similarity.[7][8]
| Aldolase-type TIM barrel | |
|---|---|
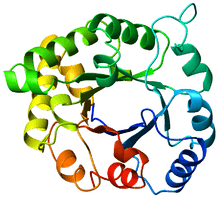 Top view of a triosephosphateisomerase (TIM) barrel (PDB: 8TIM), colored from blue (N-terminus) to red (C-terminus). | |
| Identifiers | |
| Symbol | Aldolase_TIM |
| Pfam clan | CL0036 |
| InterPro | IPR013785 |
| CATH | 8tim |
| SCOPe | 8tim / SUPFAM |
The inner β-barrel is in many cases stabilized by intricate salt-bridge networks.[9] Loops at the C-terminal ends of the β-barrel are responsible for catalytic activity[10][11] while N-terminal loops are important for the stability of the TIM-barrels. Structural inserts ranging from extended loops to independent domains may be inserted in place of these loops or at the N/C-terminals. TIM barrels appear to have evolved through gene duplication and domain fusion events of half-barrel proteins,[12] with a majority of TIM barrels originating from a common ancestor. This lead many TIM barrels to possess internal symmetries.[13] Further gene duplication events of this ancestral TIM barrel lead to diverging enzymes possessing the functional diversity observed today. TIM barrels have also been a longstanding target for protein designers. Successful TIM barrel designs include both domain fusions of existing proteins and de novo designs. Domain fusions experiments have resulted in many successful designs,[14][15][16][17][18][19][20] whereas de novo designs only yielded successes after 28 years of incremental development.[21]
Structure
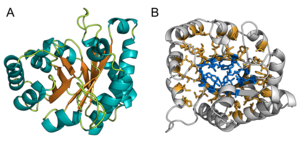
Topology
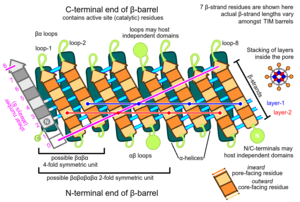
The TIM barrel gets its name from the enzyme triose phosphate isomerase (TIM), which was the first protein possessing the fold to be crystallized. TIM barrels contain 200-250 amino acid residues,[1] folded into 8 α-helices and 8 β-strands. The β-strands are arranged into a parallel β-barrel, and are surrounded by the 8 α-helices. The defining property of TIM β-barrels is that they always possess a shear number of 8.[1] The shear number is determined by picking a residue x on β-strand-1, and moving along the β-barrel, in a perpendicular direction to the direction of the strands, until residue y on the original β-strand-1 is reached. The number of residues between the start and end positions (|y−x|) is the shear number.[23] Since the number of strands is equal to the Shear number, side-chains point alternatively towards the pore and the core, giving a 4-fold symmetry. The α-helices surround and completely enclose the inner β-barrel. Short loops typically connect the α and β secondary structures, forming a (βα)8 repeat topology. In some cases, structures ranging from extended loops to independent domains may be inserted in place of these loops, or may be attached to the N/C-terminals. All TIM barrel enzymes possess catalytic sites at the C-terminal end of the β-barrel,[24] and structural inserts present close to this end may aid in catalytic activity.
Core and pore regions
TIM barrels contain two distinct buried regions, where amino acid residues are completely enveloped by their neighbors and lack access to solvent. The term 'pore' is a misnomer, as no solvent channels exist within this region. The core region consists of all residues constituting the α-β interface, and lies exterior to the central β-barrel. The pore region consists of all interior β-barrel residues, which are surrounded and enclosed by the β-barrel backbone.
Due to the pleated nature of β-strands, alternate residues along a strand are almost evenly split between the pore (53%) and core (47%). For β-barrels, 95% of their core residues are buried. Only 11% of their core residues are polar, possessing an affinity for water, and possessing the ability to form hydrogen bonds or salt bridges.[9] Similarly, 84% of β-strand pore residues are buried. However, 42% of their pore residues are polar. These residues form intricate salt bridge networks to compensate for their lack of solvent accessibility.
TIM barrel stabilizing elements
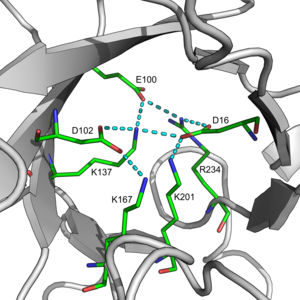
Salt bridges within TIM barrel pores are thought to contribute to the overall stability of the fold. An example of a large salt bridge network can be found in 2-deoxyribose-5-phosphate aldolase. This network was found to be conserved across the Class I aldolase family.
The exact reason for the overrepresentation of polar residues and salt bridges within the pore remains unclear. One study proposes that they improve foldabilityrather than thermodynamic stability of TIM barrels. During the folding process, inner pore residues on β-strands would be exposed to water. Partially-folded βαβα modules, called foldons, would be energetically stabilized by polar pore residues during this stage of folding.
In another study involving the S. solfataricus indole-3-glycerol phosphate synthase TIM barrel protein, a conserved βαβαβ module was found to be an essential folding template, which guided the folding of other secondary structures. β-barrel closure only occurred at the end of the folding process. In this case however, the authors credited branched aliphatic amino acids (valine, leucine, and isoleucine) for foldon stability.
Another stabilizing element in TIM barrels is the β-hairpin clamp. Side chain H-bond donors at the N-termini of even-numbered β-strands often form H-bonds with main chain amide hydrogens in preceding odd-numbered β-strands. These clamps (or hydrophobic side chain bridge analogs) are conserved in 3 indole-3-glycerolphosphate synthase TIM barrel orthologs from the bacterial and archaeal kingdoms, implying they arose in their last common ancestor and have been preserved for over a billion years.
Structural inserts
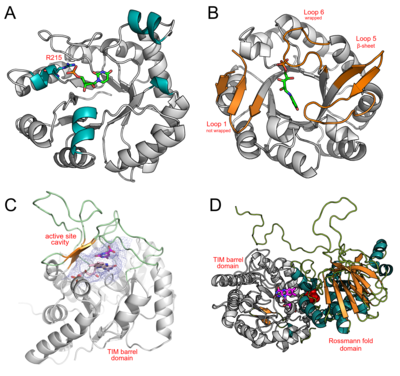
The N/C-terminal and loop regions on TIM barrel proteins are capable of hosting structural inserts ranging from simple secondary structural motifs to complete domains. These domains aid in substrate recognition and catalytic activity. Four diverse examples of TIM barrels containing additional motifs and domains are discussed below.
Bacillus subtilis Orotidine 5'-phosphate decarboxylase (PDB: 1DBT) is a TIM barrel protein displaying 4 α-helices in place of the βα loops typically present at the C-terminal of the β-barrel (residues 35-42, 89-91, 126-133, and 215-219). One of these helices (R215→K219) contains a conserved arginineresidue (R215) required for interacting with a phosphate moiety on orotidine 5′-monophosphate. The other helices were not found to host residues critical for catalytic activity, and may serve in structural roles.
Mycobacterium tuberculosis bifunctional histidine/tryptophan biosynthesis isomerase (PriA) (PDB: 2Y85) possesses the ability to catalyse two reactions: (i) HisA reaction: the conversion of N-[(5-phosphoribosyl) formimino]-5-aminoimidazole-4-carboxamide ribonucleotide (ProFAR) to N-[(5-phosphoribulosyl)formimino]-5-aminoimidazole-4-carboxamide ribonucleotide (PRFAR), and (ii) TrpF reaction: N-(5’-phosphoribosyl)-anthranilate (PRA) to 1-(O-carboxyphenylamino)- 1’-deoxyribulose-5’-phosphate (CdRP). PriA is a TIM barrel enzyme that accommodates both substrates using active site loops (loops 1, 5, and 6, extended βα loops at the C-terminal end of the β-barrel) that change conformation depending on the reactant present. Loop 1 wraps over the active site only in the presence of ProFAR. Loop5 wraps over the active site, adopting a β-sheet conformation in the presence of CdRP, or a knot-like conformation in the presence of ProFAR. Loop 6 wraps over the active site for all reactants.
Lactococcus lactis Dihydroorotate dehydrogenase A (DHODA) (PDB: 2DOR) is an example of a TIM barrel possessing β-sheets and extended loops over the C-terminal end of the β-barrel. DHODA catalyzes the oxidation of dihydroorotate to orotate, which is part of the de novo uridine 5'-monophosphate (UMP) synthesis pathway. This oxidation is mediated by flavin mononucleotide (FMN). Here, β-sheets and extended loops enclose the active site forming a cavity, while also hosting several catalytic residues.
The Methylophilus methylotrophus trimethylamine dehydrogenase (PDB: 2TMD) TIM barrel is an example of a complete domain insertion. Here, a Rossmann fold domain is inserted at the C-terminal end of the TIM-barrel. Trimethylamine dehydrogenase catalyzes the conversion of trimethylamine to formaldehyde. This reaction requires both a reduced 6-S-cysteinyl Flavin mononucleotide (FMN) cofactor and a reduced iron-sulphur ([4Fe-4S]+) center. FMN is covalently bound within the C-terminal region of the β-barrel. The [4Fe-4S]+ center is too large to be accommodated within the TIM barrel, and is instead placed in close proximity, 7 Å away, at the interface between the TIM barrel and Rossmann fold domains.
Folding mechanisms
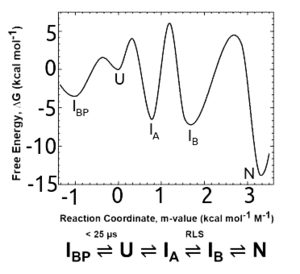
The conservation of the TIM barrel fold is mirrored by the conservation of its equilibrium and kinetic folding mechanisms in bacterial paralogs with phylogenetically distinct lineages. Chemical denaturation of several natural[26][27] and 2 designed TIM barrel variants[27] invariably involves a highly populated equilibrium intermediate. The kinetic intermediates that appear after dilution from highly denaturing solutions involve an early misfolded species that must at least partially unfold to access the productive folding pathway.[26][27] }}</ref> The rate-limiting step in folding is the closure of the 8-stranded β-barrel, with the preceding, open barrel form corresponding to the equilibrium intermediate.[28] Native-centric molecular dynamics simulations recapitulate the experimental results and point the way to testable computational models for complex folding mechanisms.[29]
Conserved fitness landscapes
TIM barrel proteins possess an unusually high sequence plasticity, forming large families of orthologous and paralogous enzymes in widely divergent organisms. This plasticity suggests a sequence landscape that allows for protein adaptation to a variety of environmental conditions, largely independent of phylogenetic history, while maintaining function. A deep mutational scanning[30] approach and a competition assay[31] was used to determine the fitness of all possible amino acid mutants across positions in 3 hyperthermophilic indole-3-glycerolphosphate synthase (IGPS) TIM barrel enzymes in supporting the growth of a yeast host lacking IGPS. Although the 2 bacterial and 1 archaeal IGPS enzymes were only 30-40% identical in sequence, their fitness landscapes were strongly correlated: the same amino acids at the same positions in the three different proteins had very similar fitness. The correlation can be thought of as the conservation of the fitness landscape for a TIM barrel enzyme across evolutionary time.
Loop regions
Of the approximately 200 residues required to fully form a TIM barrel, about 160 are considered structurally equivalent between different proteins sharing this fold. The remaining residues are located on the loop regions that link the helices and strands; the loops at the C-terminal end of the strands tend to contain the active site, which is one reason this fold is so common: the residues required to maintain the structure and the residues that effect enzymatic catalysis are for the most part distinct subsets:[32] The linking loops can, in fact, be so long that they contain other protein domains. Recently, it has been demonstrated that catalytic loops can be exchanged between different TIM barrel enzymes as semiautonomous units of functional groups.[33]
Evolution and origins
The predominant theory for TIM barrel evolution involves gene duplication and fusion, starting with a half- barrel that eventually formed a full TIM barrel. Multiple studies support the theory of divergent evolution from a single ancestor, and are discussed below.
Evolution from a common ancestor
In the early 1990s, it was noted that all TIM barrel structures solved at the time were enzymes, indicating divergence from a common ancestor.[10][11] Further, all TIM barrels possessed active sites at the C-terminal end of β-barrels. suggested that A common phosphate binding site, formed by a small α-helix and TIM barrel loops-7/8, strongly indicated divergent evolution.[34] Further studies of these phosphate groups, concluding that 12 of 23 SCOP TIM barrel families diverged from a common ancestor.[35] Similarly there were hints for common ancestry for 17 of the 21 CATH TIM barrel families.[6] Based on these reports, it is considered plausible that the majority of TIM barrel proteins evolved from a common ancestor.
Origin through gene duplication and domain fusion

Many TIM barrel proteins possess 2-fold, 4-fold or 8-fold internal symmetry, suggesting that TIM barrels evolved from ancestral (βα)4, (βα)2, or βα motifs through gene duplication and domain fusion. A good example of 2-fold internal symmetry is observed in the enzymes ProFAR isomerase (HisA) and imidazole glycerol phosphate synthase (HisF) of the Thermotoga maritima histidine biosynthesis pathway.[12] They catalyze 2 successive reactions in the pathway, possess 25% sequence homology, and possess root-mean-square deviations (RMSDs) between 1.5-2 Å, suggesting divergence from a common ancestor. More interestingly, the loops on the C terminal ends of both HisA and HisF showed a twofold repeated pattern, suggesting that their common ancestor also possessed 2-fold internal symmetry. Using these observations, a model was constructed for the evolution of the TIM barrels.[12] An ancestral half-barrel would have undergone a gene duplication and fusion event, resulting in a single protein containing two half-barrel domains. Structural adaptations would have occurred, resulting in the merging of these domains to form a closed β-barrel, and forming an ancestral TIM barrel. Functional adaptations would have also occurred, resulting in the evolution of new catalytic activity at the C terminal end of the β-barrel. At this point, the common ancestor of HisA and HisF would have undergone a second gene duplication event. Divergent evolution of the duplicated genes of the ancestral TIM barrel would have resulted in the formation of HisA and HisF.
Interestingly, this evolutionary model has been experimentally validated using rational protein design and directed evolution. Höcker et al. first fused two C-terminal halves of HisF, yielding HisF-CC. This construct was then stabilized by the insertion of an internal salt-bridge, yielding HisF-C*C.[16] Further stepwise stabilization and solubilization of HisF-C*C was achieved by optimizing the half-barrel interface, generating HisF-C**C and HisF-C***C, respectively.[14][15] The crystal structure of HisF-C***C revealed a 2-fold symmetric TIM barrel, validating the possibility of natural domain fusion. Moreover, Höcker created the first chimeric HisAF and HisFA TIM barrels using HisA and HisF half-barrels.[16] These experiments led to the proposal of a novel means of diversification and evolution of TIM-barrel enzymes through the exchange of (βα)4 half-barrel domains amongst preexisting TIM barrels. In accordance with this idea, a high catalytic activity on the HisAF construct was established.[17] Similarly, chimeric βα5-flavodoxin-like fold (CheY)/HisF TIM barrels,[18][19] and a perfectly 2-fold symmetric HisF-based TIM barrel[20][27] have also been created.
The existence of 4/8-fold internal symmetry was suggested based on a computational analysis of TIM barrel sequences.[13] For example, Escherichia coli KDPG aldolase[36] (PDB: 1FQ0) was suggested to possess a distinct 4-fold symmetry, with discernible 8-fold symmetry. The design of a 4-fold symmetric TIM barrel[21] confirmed the possibility of higher orders of internal symmetry in natural TIM barrels, and will be discussed in detail in the next section. No experimental evidence for the existence of 8-fold symmetric TIM barrels has been reported to date.
De novo TIM barrel design
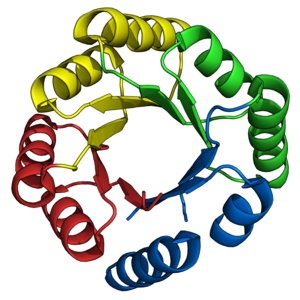
The TIM barrel fold has been a long-standing target for de novo protein designers. As previously described, numerous TIM barrels have been successfully designed based on preexisting natural half-barrels. In contrast, the de novo design of TIM barrels occurred in incremental steps over a period of 28 years.[37]
The Octarellin series[38][39][40][41][42] of proteins (Octarellin I→VI) were the first attempts to create a de novo TIM barrel. As the field of protein design was still in its infancy, these design attempts were only met with limited success. Although they displayed circular dichroism spectra consistent with αβ proteins and some cooperative folding characteristics, all Octarellin series peptides were insoluble, and had to be resolubilized from inclusion bodies for further characterization. Interestingly, Octarellin V.1[43] displayed a Rossmann-like fold under co-crystal conditions.
The Symmetrin series of proteins (Symmetrin-1→4) displayed more favorable biophysical characteristics. Symmetrin-1 was readily soluble, displayed circular dichroism spectra consistent with αβ proteins, and displayed excellent cooperative unfolding and refolding characteristics. Despite these advances, all proteins in this family displayed molten characteristics when analyzed using NMR (nuclear magnetic resonance), and further work to solve their structures could not be pursued.
Proteins of the sTIM series[21] represented the first successful de novo TIM barrel design.[44][37] sTIM-11 (PDB: 5BVL) was designed with an internal 4-fold symmetry, to reduce the complexity of computational design using the Rosetta software suite.[45] Previously-derived first principles[46] were used to delineate secondary structure topologies and lengths. sTIM-11 proved to be a highly thermostable, cooperatively folding design that adopted its intended structure.
See also
References
![]()
- Wierenga RK (March 2001). "The TIM-barrel fold: a versatile framework for efficient enzymes". FEBS Letters. 492 (3): 193–8. doi:10.1016/s0014-5793(01)02236-0. PMID 11257493.
- Jansen R, Gerstein M (March 2000). "Analysis of the yeast transcriptome with structural and functional categories: characterizing highly expressed proteins". Nucleic Acids Research. 28 (6): 1481–8. doi:10.1093/nar/28.6.1481. PMC 111042. PMID 10684945.
- Nagano N, Hutchinson EG, Thornton JM (October 1999). "Barrel structures in proteins: automatic identification and classification including a sequence analysis of TIM barrels". Protein Science. 8 (10): 2072–84. doi:10.1110/ps.8.10.2072. PMC 2144152. PMID 10548053.
- Webb, Edwin C. (1992). Enzyme nomenclature: Recommendations of the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology on the Nomenclature and Classification of Enzymes. Academic Press. ISBN 978-0-12-227164-9.
- Nagano N, Orengo CA, Thornton JM (August 2002). "One fold with many functions: the evolutionary relationships between TIM barrel families based on their sequences, structures and functions". Journal of Molecular Biology. 321 (5): 741–65. doi:10.1016/s0022-2836(02)00649-6. PMID 12206759.
- Livesay DR, La D (May 2005). "The evolutionary origins and catalytic importance of conserved electrostatic networks within TIM-barrel proteins". Protein Science. 14 (5): 1158–70. doi:10.1110/ps.041221105. PMC 2253277. PMID 15840824.
- Chung SY, Subbiah S (October 1996). "A structural explanation for the twilight zone of protein sequence homology". Structure. 4 (10): 1123–7. doi:10.1016/s0969-2126(96)00119-0. PMID 8939745.
- Vijayabaskar MS, Vishveshwara S (2012). "Insights into the fold organization of TIM barrel from interaction energy based structure networks". PLOS Computational Biology. 8 (5): e1002505. doi:10.1371/journal.pcbi.1002505. PMC 3355060. PMID 22615547.
- Farber GK, Petsko GA (June 1990). "The evolution of alpha/beta barrel enzymes". Trends in Biochemical Sciences. 15 (6): 228–34. doi:10.1016/0968-0004(90)90035-A. PMID 2200166.
- Reardon D, Farber GK (April 1995). "The structure and evolution of alpha/beta barrel proteins". FASEB Journal. 9 (7): 497–503. doi:10.1096/fasebj.9.7.7737457. PMID 7737457.
- Lang D, Thoma R, Henn-Sax M, Sterner R, Wilmanns M (September 2000). "Structural evidence for evolution of the beta/alpha barrel scaffold by gene duplication and fusion". Science. 289 (5484): 1546–50. doi:10.1126/science.289.5484.1546. PMID 10968789.
- Söding J, Remmert M, Biegert A (July 2006). "HHrep: de novo protein repeat detection and the origin of TIM barrels". Nucleic Acids Research. 34 (Web Server issue): W137-42. doi:10.1093/nar/gkl130. PMC 1538828. PMID 16844977.
- Seitz T, Bocola M, Claren J, Sterner R (September 2007). "Stabilisation of a (betaalpha)8-barrel protein designed from identical half barrels". Journal of Molecular Biology. 372 (1): 114–29. doi:10.1016/j.jmb.2007.06.036. PMID 17631894.
- Höcker B, Lochner A, Seitz T, Claren J, Sterner R (February 2009). "High-resolution crystal structure of an artificial (betaalpha)(8)-barrel protein designed from identical half-barrels". Biochemistry. 48 (6): 1145–7. doi:10.1021/bi802125b. PMID 19166324.
- Höcker B, Claren J, Sterner R, Makar AB, McMartin KE, Palese M, Tephly TR (June 1975). "Formate assay in body fluids: application in methanol poisoning". Biochemical Medicine. 13 (2): 117–26. doi:10.1016/0006-2944(75)90147-7. PMC 534502. PMID 15539462.
- Claren J, Malisi C, Höcker B, Sterner R (March 2009). "Establishing wild-type levels of catalytic activity on natural and artificial (beta alpha)8-barrel protein scaffolds". Proceedings of the National Academy of Sciences of the United States of America. 106 (10): 3704–9. doi:10.1073/pnas.0810342106. PMC 2656144. PMID 19237570.
- Bharat TA, Eisenbeis S, Zeth K, Höcker B (July 2008). "A beta alpha-barrel built by the combination of fragments from different folds". Proceedings of the National Academy of Sciences of the United States of America. 105 (29): 9942–7. doi:10.1073/pnas.0802202105. PMC 2481348. PMID 18632584.
- Eisenbeis S, Proffitt W, Coles M, Truffault V, Shanmugaratnam S, Meiler J, Höcker B (March 2012). "Potential of fragment recombination for rational design of proteins". Journal of the American Chemical Society. 134 (9): 4019–22. doi:10.1021/ja211657k. PMID 22329686.
- Fortenberry C, Bowman EA, Proffitt W, Dorr B, Combs S, Harp J, et al. (November 2011). "Exploring symmetry as an avenue to the computational design of large protein domains". Journal of the American Chemical Society. 133 (45): 18026–9. doi:10.1021/ja210593m. PMC 3781211. PMID 21978247.
- Huang PS, Feldmeier K, Parmeggiani F, Velasco DA, Höcker B, Baker D (January 2016). "De novo design of a four-fold symmetric TIM-barrel protein with atomic-level accuracy". Nature Chemical Biology. 12 (1): 29–34. doi:10.1038/nchembio.1966. PMC 4684731. PMID 26595462.
- Nagarajan D, Deka G, Rao M (August 2015). "Design of symmetric TIM barrel proteins from first principles". BMC Biochemistry. 16 (1): 18. doi:10.1186/s12858-015-0047-4. PMC 4531894. PMID 26264284.
- Murzin AG, Lesk AM, Chothia C (March 1994). "Principles determining the structure of beta-sheet barrels in proteins. I. A theoretical analysis". Journal of Molecular Biology. 236 (5): 1369–81. doi:10.1016/0022-2836(94)90064-7. PMID 8126726.
- Brändén C (1991). "The TIM barrel—the most frequently occurring folding motif in proteins". Current Opinion in Structural Biology. 1 (6): 978–983. doi:10.1016/0959-440x(91)90094-a.
- Barber MJ, Neame PJ, Lim LW, White S, Matthews FS (April 1992). "Correlation of x-ray deduced and experimental amino acid sequences of trimethylamine dehydrogenase". The Journal of Biological Chemistry. 267 (10): 6611–9. PMID 1551870.
- Forsyth WR, Bilsel O, Gu Z, Matthews CR (September 2007). "Topology and sequence in the folding of a TIM barrel protein: global analysis highlights partitioning between transient off-pathway and stable on-pathway folding intermediates in the complex folding mechanism of a (betaalpha)8 barrel of unknown function from B. subtilis". Journal of Molecular Biology. 372 (1): 236–53. doi:10.1016/j.jmb.2007.06.018. PMID 17619021.
- Carstensen L, Sperl JM, Bocola M, List F, Schmid FX, Sterner R (August 2012). "Conservation of the folding mechanism between designed primordial (βα)8-barrel proteins and their modern descendant". Journal of the American Chemical Society. 134 (30): 12786–91. doi:10.1021/ja304951v. PMID 22758610.
- Gu Z, Rao MK, Forsyth WR, Finke JM, Matthews CR (November 2007). "Structural analysis of kinetic folding intermediates for a TIM barrel protein, indole-3-glycerol phosphate synthase, by hydrogen exchange mass spectrometry and Gō model simulation". Journal of Molecular Biology. 374 (2): 528–46. doi:10.1016/j.jmb.2007.09.024. PMC 2735044. PMID 17942114.
- Halloran KT, Wang Y, Arora K, Chakravarthy S, Irving TC, Bilsel O, et al. (August 2019). "Frustration and folding of a TIM barrel protein". Proceedings of the National Academy of Sciences of the United States of America. 116 (33): 16378–16383. doi:10.1073/pnas.1900880116. PMC 6697809. PMID 31346089.
- Chan YH, Venev SV, Zeldovich KB, Matthews CR (March 2017). "Correlation of fitness landscapes from three orthologous TIM barrels originates from sequence and structure constraints". Nature Communications. 8: 14614. doi:10.1038/ncomms14614. PMC 5343507. PMID 28262665.
- Hietpas RT, Jensen JD, Bolon DN (May 2011). "Experimental illumination of a fitness landscape". Proceedings of the National Academy of Sciences of the United States of America. 108 (19): 7896–901. doi:10.1073/pnas.1016024108. PMC 3093508. PMID 21464309.
- Ochoa-Leyva A, Soberón X, Sánchez F, Argüello M, Montero-Morán G, Saab-Rincón G (April 2009). "Protein design through systematic catalytic loop exchange in the (beta/alpha)8 fold". Journal of Molecular Biology. 387 (4): 949–64. doi:10.1016/j.jmb.2009.02.022. PMID 19233201.
- Ochoa-Leyva A, Barona-Gómez F, Saab-Rincón G, Verdel-Aranda K, Sánchez F, Soberón X (August 2011). "Exploring the Structure-Function Loop Adaptability of a (β/α)(8)-Barrel Enzyme through Loop Swapping and Hinge Variability". Journal of Molecular Biology. 411 (1): 143–57. doi:10.1016/j.jmb.2011.05.027. PMID 21635898.
- Brändén, Carl-Ivar (1991). "The TIM barrel—the most frequently occurring folding motif in proteins". Current Opinion in Structural Biology. 1 (6): 978–983. doi:10.1016/0959-440x(91)90094-a.
- Copley RR, Bork P (November 2000). "Homology among (betaalpha)(8) barrels: implications for the evolution of metabolic pathways". Journal of Molecular Biology. 303 (4): 627–41. doi:10.1006/jmbi.2000.4152. PMID 11054297.
- Wymer N, Buchanan LV, Henderson D, Mehta N, Botting CH, Pocivavsek L, et al. (January 2001). "Directed evolution of a new catalytic site in 2-keto-3-deoxy-6-phosphogluconate aldolase from Escherichia coli". Structure. 9 (1): 1–9. doi:10.1016/S0969-2126(00)00555-4. PMID 11342129.
- Borman S (2015). "Protein designers roll out a barrel". Chemical & Engineering News. 93 (47). p. 6.
- Goraj K, Renard A, Martial JA (March 1990). "Synthesis, purification and initial structural characterization of octarellin, a de novo polypeptide modelled on the alpha/beta-barrel proteins". Protein Engineering. 3 (4): 259–66. doi:10.1093/protein/3.4.259. PMID 2188263.
- Beauregard M, Goraj K, Goffin V, Heremans K, Goormaghtigh E, Ruysschaert JM, Martial JA (October 1991). "Spectroscopic investigation of structure in octarellin (a de novo protein designed to adopt the alpha/beta-barrel packing)". Protein Engineering. 4 (7): 745–9. doi:10.1093/protein/4.7.745. PMID 1798699.
- Houbrechts A, Moreau B, Abagyan R, Mainfroid V, Préaux G, Lamproye A, et al. (March 1995). "Second-generation octarellins: two new de novo (beta/alpha)8 polypeptides designed for investigating the influence of beta-residue packing on the alpha/beta-barrel structure stability". Protein Engineering. 8 (3): 249–59. doi:10.1093/protein/8.3.249. PMID 7479687.
- Offredi F, Dubail F, Kischel P, Sarinski K, Stern AS, Van de Weerdt C, et al. (January 2003). "De novo backbone and sequence design of an idealized alpha/beta-barrel protein: evidence of stable tertiary structure". Journal of Molecular Biology. 325 (1): 163–74. doi:10.1016/S0022-2836(02)01206-8. PMID 12473459.
- Figueroa M, Oliveira N, Lejeune A, Kaufmann KW, Dorr BM, Matagne A, et al. (2013). "Octarellin VI: using rosetta to design a putative artificial (β/α)8 protein". PLOS ONE. 8 (8): e71858. doi:10.1371/journal.pone.0071858. PMC 3747059. PMID 23977165.
- Figueroa M, Sleutel M, Vandevenne M, Parvizi G, Attout S, Jacquin O, et al. (July 2016). "The unexpected structure of the designed protein Octarellin V.1 forms a challenge for protein structure prediction tools". Journal of Structural Biology. 195 (1): 19–30. doi:10.1016/j.jsb.2016.05.004. PMID 27181418.
- Nanda V (January 2016). "Protein Design: Getting to the bottom of the TIM barrel". Nature Chemical Biology. 12 (1): 2–3. doi:10.1038/nchembio.1987. PMID 26678608.
- Kaufmann KW, Lemmon GH, Deluca SL, Sheehan JH, Meiler J (April 2010). "Practically useful: what the Rosetta protein modeling suite can do for you". Biochemistry. 49 (14): 2987–98. doi:10.1021/bi902153g. PMC 2850155. PMID 20235548.
- Koga N, Tatsumi-Koga R, Liu G, Xiao R, Acton TB, Montelione GT, Baker D (November 2012). "Principles for designing ideal protein structures". Nature. 491 (7423): 222–7. doi:10.1038/nature11600. PMC 3705962. PMID 23135467.
External links
- SCOP list of proteins adopting the TIM barrel fold
- Babu MM (1998). "TIM Barrel Analysis". Center for Biotechnology, Anna University.