Roberts syndrome
Roberts syndrome, or sometimes called pseudothalidomide syndrome, is an extremely rare autosomal recessive genetic disorder that is characterized by mild to severe prenatal retardation or disruption of cell division, leading to malformation of the bones in the skull, face, arms, and legs.
| Roberts syndrome | |
|---|---|
| Other names | Hypomelia-hypotrichosis-facial hemangioma syndrome, SC syndrome (once thought to be an entirely separate disease), pseudothalidomide syndrome, Roberts-SC phocomelia syndrome, SC phocomelia syndrome, Appelt-Gerken-Lenz syndrome, RBS, SC pseudothalidomide syndrome, and tetraphocomelia-cleft palate syndrome.[1][2][3][4] |
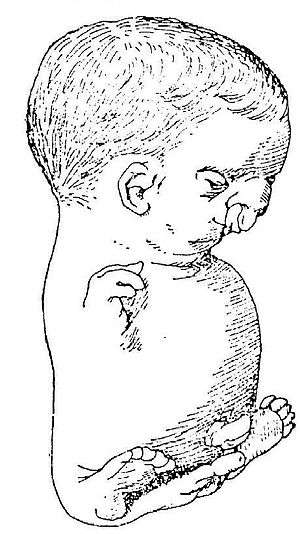 | |
| Specialty | Medical genetics |
It is caused by a mutation in the ESCO2 gene. It is one of the rarest autosomal recessive disorders, affecting approximately 150 known individuals. The mutation causes cell division to occur slowly or unevenly, and the cells with abnormal genetic content die.
Roberts syndrome can affect both males and females. Although the disorder is rare, the affected group is diverse. The mortality rate is high in severely affected individuals. The syndrome is named after American surgeon and physician John Bingham Roberts (1852–1924), who first described it in 1919.
Symptoms
The following is a list of symptoms that have been associated with Roberts syndrome:
- Bilateral symmetric tetraphocomelia- a birth defect in which the hands and feet are attached to shortened arms and legs
- Prenatal growth retardation
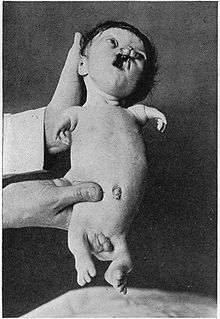
 An example of a severely affected Roberts syndrome Patient
An example of a severely affected Roberts syndrome Patient - Hypomelia (hypoplasia)- the incomplete development of a tissue or organ; less drastic than aplasia, which is no development at all
- Oligodactyly- fewer than normal number of fingers or toes
- Thumb aplasia- the absence of a thumb
- Syndactyly- condition in which two or more fingers (or toes) are joined together; the joining can involve the bones or just the skin between the fingers
- Clinodactyly- curving of the fifth finger (little finger) towards the fourth finger (ring finger) due to the underdevelopment of the middle bone in the fifth finger
- Elbow/knee flexion contractures- an inability to fully straighten the arm or leg
- Cleft lip- the presence of one or two vertical fissures in the upper lip; can be on one side (unilateral) or on both sides (bilateral)
- Cleft palate- opening in the roof of the mouth
- Premaxillary protrusion- upper part of the mouth sticks out farther than the lower part of the mouth
- Micrognathia- small chin
- Microbrachycephaly- smaller than normal head size
- Malar hypoplasia- underdevelopment of the cheek bones
- Downslanting palpebral fissures- the outer corners of the eyes point downwards
- Ocular hypertelorism- unusually wide-set eyes
- Exophthalmos- a protruding eyeball
- Corneal clouding- clouding of the front-most part of the eye
- Hypoplastic nasal alae- narrowing of the nostrils that can decrease the width of the nasal base
- Beaked nose- a nose with a prominent bridge that gives it the appearance of being curved
- Ear malformations
- Intellectual disability
- Encephalocele (only in severe cases)- rare defect of the neural tube characterized by sac-like protrusions of the brain
Mortality is high among those severely affected by Roberts syndrome; however, mildly affected individuals may survive to adulthood[1][3][4]
Heredity

From Hirst & Piersol, 1893
ESCO2, located on human chromosome 8, has been labeled as the gene responsible for Roberts syndrome. In fact, ESCO2 is the only known gene that has demonstrated RBS-causing mutations. Also, all individuals that have been cytogenetically diagnosed with Roberts syndrome have also had mutations in the ESCO2 gene.[3]
In order to contract Roberts syndrome, a child must inherit the defective gene in an autosomal recessive manner. In other words, the child must inherit two copies of the defective gene (one from each parent). The ESCO2 gene has a specific effect on cell division in Roberts syndrome patients. In normal cell division, each chromosome is copied and then attached to its newly formed copy at the centromere (the center portion of a chromosome). However, in Roberts syndrome cell division, the copies are frequently not attached at the centromere. As a result, the chromosomes do not get lined up properly, which causes the cell to divide very slowly or even to not divide at all. The new cells typically will have too many or too few chromosomes. The odd number of chromosomes causes the defective cells to die, which leads to the malformations associated with Roberts syndrome.[1]
Many of the physical malformations associated with Roberts syndrome are very similar to the malformations that occur in children whose mothers took thalidomide during pregnancy. The physical similarities suggest that there is a similar underlying biology between ESCO2 and thalidomide. As a result, it is speculated that thalidomide affects chromosomes and cell division in a similar manner to ESCO2. For this reason, Roberts syndrome is sometimes called Pseudothalidomide Syndrome.
Discovery of the syndrome
The discovery of ESCO2 as the gene responsible for Roberts syndrome was made by studying samples from fifteen families affected by Roberts syndrome. In 1995, Hugo Vega and Miriam Gordillo, two Colombian geneticists, set out to fully understand Roberts syndrome. Vega and Gordillo noticed an unusually high number of Roberts syndrome patients at the Universidad Nacional de Colombia. The two Colombian geneticists tracked down a total of seven families with Roberts syndrome just outside Bogota and discovered that four out of the seven families shared a common 18th century ancestor. Using this information, Vega and Gordillo were able to pinpoint the gene responsible for Roberts syndrome, which was ESCO2.[5]
Diagnosis
Clinical diagnosis
A clinical diagnosis of Roberts syndrome is made in individuals with characteristic prenatal growth retardation, limb malformations, and craniofacial abnormalities. The specific characteristics that are looked for in the clinical diagnosis are listed below.
- Prenatal growth retardation- low birth length and weight that can range from mild to severe
- Limb malformations- bilateral symmetric tetraphocomelia, oligodactyly, thumb aplasia, syndactyly, clinodactyly, and elbow and knee flexion contractures
- Craniofacial abnormalities- bilateral cleft lip and palate, micrognathia, hypertelorism, exophthalmos, down-slanting palpebral fissures, malar hypoplasia, hypoplastic nasal alae, and ear malformations
An official diagnosis of Roberts syndrome relies on cytogenetic testing of the peripheral blood.[6]
Testing
Cytogenetic testing
Cytogenetic preparations that have been stained by either Giemsa or C-banding techniques will show two characteristic chromosomal abnormalities. The first chromosomal abnormality is called premature centromere separation (PCS) and is the most likely pathogenic mechanism for Roberts syndrome. Chromosomes that have PCS will have their centromeres separate during metaphase rather than anaphase (one phase earlier than normal chromosomes). The second chromosomal abnormality is called heterochromatin repulsion (HR). Chromosomes that have HR experience separation of the heterochromatic regions during metaphase. Chromosomes with these two abnormalities will display a "railroad track" appearance because of the absence of primary constriction and repulsion at the heterochromatic regions. The heterochromatic regions are the areas near the centromeres and nucleolar organizers. Carrier status cannot be determined by cytogenetic testing. Other common findings of cytogenetic testing on Roberts syndrome patients are listed below.
- Aneuploidy- the occurrence of one or more extra or missing chromosomes
- Micronucleation- nucleus is smaller than normal
- Multilobulated nuclei- the nucleus has more than one lobe[6]
Genetic testing
At this point in time, ESCO2 is the only known gene to cause Roberts syndrome mutations. Also, all individuals that have been diagnosed with Roberts syndrome by cytogenetic techniques have also had ESCO2 mutations. Confirmation of a Roberts syndrome diagnosis requires detection of the characteristic chromosomal abnormalities (PCS and HR) or the identification of two ESCO2 mutations that have been linked to Roberts syndrome.[6]
Carrier testing and prenatal diagnosis
Carrier testing for Roberts syndrome requires prior identification of the disease-causing mutation in the family. Carriers for the disorder are heterozygotes due to the autosomal recessive nature of the disease. Carriers are also not at risk for contracting Roberts syndrome themselves. A prenatal diagnosis of Roberts syndrome requires an ultrasound examination paired with cytogenetic testing or prior identification of the disease-causing ESCO2 mutations in the family.[6]
Genetically related disorders
At this time, there are no other phenotypes (observable expressions of a gene) that have been discovered for mutations in the ESCO2 gene.[6]
Differential diagnosis
In cases of mild malformations, the following disorders should be considered in the differential diagnosis:
- Baller-Gerold Syndrome
- Fanconi Anemia (FA)
In cases of severe manifestations, the following disorders should be considered in the differential diagnosis:
- Thrombocytopenia-Absent Radius (TAR) Syndrome
- Tetra-Amelia, X-linked
- Tetra-Amelia, Autosomal Recessive
- Splenogonadal Fusion with Limb Defects and Micrognathia
- DK Phocomelia Syndrome
- Holt-Oram Syndrome
- Thalidomide Embryopathy
In cases of similar cytogenetic findings, the following disorders should be considered in the differential diagnosis:
- Cornelia de Lange Syndrome (CdLS)
- Mosaic Variegated Aneuploidy Syndrome[6]
Clinical description
Little is known about the natural history of Roberts syndrome due to its wide clinical variability. The prognosis of the disease depends on the malformations, as the severity of the malformations correlates with survival. The cause of death for most fatalities of Roberts syndrome have not been reported; however, five deaths were reportedly due to infection.
The following are observations that have been made in individuals with cytogenetic findings of PCS/HR or ESCO2 mutations:
- The symptom of prenatal growth retardation is the most common finding and can be moderate to severe. Postnatal growth retardation can also be moderate to severe and correlates with the degree of severity of limb and craniofacial malformations.
- In limb malformations, the upper limbs are typically more severely affected than the lower limbs. There have been many cases of only upper limb malformation.
- In hand malformations, the thumb is most often affected, followed by the fifth finger (the little finger). In severe cases, the patient may only have three fingers and in rare cases only one.
- In craniofacial malformations, mildly affected individuals will have no abnormalities of the palate. The most severely affected will have a fronto-ethmoid-nasal-maxillary encephalocele.
- The severity of limb malformations and craniofacial malformations is correlated.
- Other abnormalities can occur in different parts of the body, including:
- Heart- atrial septal defects, ventricular septal defects, patent ductus arteriosus
- Kidneys- polycystic kidney, horseshoe kidney
- Male genitals- enlarged penis, cryptorchidism
- Female genitals- enlarged clitoris
- Hair- sparse, silvery-blonde scalp hair
- Cranial nerve paralysis, moyamoya disease, stroke, intellectual disability[3]
Treatment
Treatment of Roberts syndrome is individualized and specifically aimed at improving the quality of life for those afflicted with the disorder. Some of the possible treatments include: surgery for the cleft lip and palate, correction of limb abnormalities (also through surgery), and improvement in prehensile hand grasp development.[3]
Prevalence
Roberts syndrome is an extremely rare condition that only affects about 150 reported individuals. Although there have been only about 150 reported cases, the affected group is quite diverse and spread worldwide. Parental consanguinity (parents are closely related) is common with this genetic disorder. The frequency of Roberts syndrome carriers is unknown.[3][4]
Nomenclature
Roberts Syndrome is named after Dr. John Bingham Roberts (1852–1924) of Philadelphia, who reported the disease characteristics in 1919. Roberts reported a disease that was characterized by phocomelia, cleft lip, cleft palate, and a protrusion of the intermaxillary region in three siblings of an Italian couple. The Italian couple were first cousins, which made Roberts Syndrome acquisition more likely for their children due to the diseases autosomal recessive nature.
Later, in 1969, J. Herrmann described another syndrome with very similar characteristics to Roberts syndrome. Herrmann would call the disorder Pseudothalidomide Syndrome or SC Syndrome (SC was for the initials of the surnames of the two families that Herrmann studied). Today, Roberts Syndrome and Pseudothalidomide Syndrome (SC Syndrome) are considered to be the same disorder.
The following is a list of all the alternate names that have been used for Roberts Syndrome:
- RBS
- Hypomelia-Hypotrichosis-Facial Hemangioma Syndrome
- SC Syndrome
- Pseudothalidomide Syndrome
- Roberts-SC Phocomelia Syndrome
- SC Phocomelia Syndrome
- Appelt-Gerken-Lenz Syndrome
- SC Pseudothalidomide Syndrome
- Tetraphocomelia-Cleft Palate Syndrome[2][3][4]
References
- Kugler, Mary. "Roberts syndrome: Inherited Disorder Causes Abnormal Bone Development." About.com: Rare Diseases. Published 23 April 2005. Accessed 13 March 2010
- Francke, Uta, and Jinglan Liu. "Roberts syndrome." National Organization for Rare Disorders. Published 26 November 2008.
- Gordillo et al. "Roberts syndrome."
- "Roberts syndrome." Genetics Home Reference. 2010. U.S. National Library of Medicine. 13 March 2010.
- Downer, Joanna."Fifteen Year Hunt Uncovers Gene Behind 'Pseudothalidomide' Syndrome." Press Releases. Johns Hopkins Medicine. 11 April 2005.
- Gordillo, Miriam, and Hugo Vega, and Ethylin Wang Jabs. "Roberts syndrome." GeneReviews. 2009. University of Washington, Seattle. 13 March 2010.
- "Transactions of the Philadelphia Academy of Surgery: Stated Meeting held May 5, 1919". Annals of Surgery. 70 (2): 251–4. 1919. doi:10.1097/00000658-191908000-00019. PMC 1410314. PMID 17864157.
Further reading
- Kugler, Mary. "Roberts syndrome: Inherited Disorder Causes Abnormal Bone Development." About.com: Rare Diseases. About. 23 April 2005.
- Downer, Joanna. "Fifteen Year Hunt Uncovers Gene Behind 'Pseudothalidomide' Syndrome." Press Releases. Johns Hopkins Medicine. 11 April 2005.
- Francke, Uta, and Jinglan Liu. "Roberts syndrome." National Organization for Rare Disorders. 26 November 2008.
- Gordillo, Miriam, and Hugo Vega, and Ethylin Wang Jabs. "Roberts syndrome." GeneReviews. 2009. University of Washington, Seattle. 13 March 2010.
- "Roberts syndrome." Genetics Home Reference. 2010. U.S. National Library of Medicine. Accessed 13 March 2010.
- "Roberts syndrome." WebMD. 2009. 13 March 2010.
- Silva, Sandra, and Philippe Jeanty. Roberts syndrome. . 1999. SonoWorld. 13 March 2010.
- "NINDS Encephaloceles Information Page." National Institute of Neurological Disorders and Stroke. 2007. National Institutes of Health. 13 March 2010.
External links
| Classification | |
|---|---|
| External resources |
Chromosome+disorders at the US National Library of Medicine Medical Subject Headings (MeSH)