Nucleophilic acyl substitution
Nucleophilic acyl substitution describe a class of substitution reactions involving nucleophiles and acyl compounds. In this type of reaction, a nucleophile – such as an alcohol, amine, or enolate – displaces the leaving group of an acyl derivative – such as an acid halide, anhydride, or ester. The resulting product is a carbonyl-containing compound in which the nucleophile has taken the place of the leaving group present in the original acyl derivative. Because acyl derivatives react with a wide variety of nucleophiles, and because the product can depend on the particular type of acyl derivative and nucleophile involved, nucleophilic acyl substitution reactions can be used to synthesize a variety of different products.
Reaction mechanism
Carbonyl compounds react with nucleophiles via an addition mechanism: the nucleophile attacks the carbonyl carbon, forming a tetrahedral intermediate. This reaction can be accelerated by acidic conditions, which make the carbonyl more electrophilic, or basic conditions, which provide a more anionic and therefore more reactive nucleophile. The tetrahedral intermediate itself can be an alcohol or alkoxide, depending on the pH of the reaction.
The tetrahedral intermediate of an acyl compound contains a substituent attached to the central carbon that can act as a leaving group. After the tetrahedral intermediate forms, it collapses, recreating the carbonyl C=O bond and ejecting the leaving group in an elimination reaction. As a result of this two-step addition/elimination process, the nucleophile takes the place of the leaving group on the carbonyl compound by way of an intermediate state that does not contain a carbonyl. Both steps are reversible and as a result, nucleophilic acyl substitution reactions are equilibrium processes.[1] Because the equilibrium will favor the product containing the best nucleophile, the leaving group must be a comparatively poor nucleophile in order for a reaction to be practical.
Acidic conditions
Under acidic conditions, the carbonyl group of the acyl compound 1 is protonated, which activates it towards nucleophilic attack. In the second step, the protonated carbonyl 2 is attacked by a nucleophile (H−Z) to give tetrahedral intermediate 3. Proton transfer from the nucleophile (Z) to the leaving group (X) gives 4, which then collapses to eject the protonated leaving group (H−X), giving protonated carbonyl compound 5. The loss of a proton gives the substitution product, 6. Because the last step involves the loss of a proton, nucleophilic acyl substitution reactions are considered catalytic in acid. Also note that under acidic conditions, a nucleophile will typically exist in its protonated form (i.e. H−Z instead of Z−).

Basic conditions
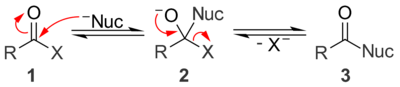
Under basic conditions, a nucleophile (Nuc) attacks the carbonyl group of the acyl compound 1 to give tetrahedral alkoxide intermediate 2. The intermediate collapses and expels the leaving group (X) to give the substitution product 3. While nucleophilic acyl substitution reactions can be base-catalyzed, the reaction will not occur if the leaving group is a stronger base than the nucleophile (i.e. the leaving group must have a higher pKa than the nucleophile). Unlike acid-catalyzed processes, both the nucleophile and the leaving group exist as anions under basic conditions.
This mechanism is supported by isotope labeling experiments. When ethyl propionate with an oxygen-18-labeled ethoxy group is treated with sodium hydroxide (NaOH), the oxygen-18 label is completely absent from propionic acid and is found exclusively in the ethanol.[2]

Reactivity trends
There are five main types of acyl derivatives. Acid halides are the most reactive towards nucleophiles, followed by anhydrides, esters, and amides. Carboxylate ions are essentially unreactive towards nucleophilic substitution, since they possess no leaving group. The reactivity of these five classes of compounds covers a broad range; the relative reaction rates of acid chlorides and amides differ by a factor of 1013.[3]
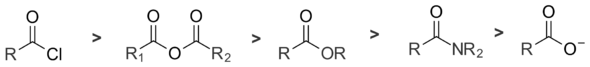
A major factor in determining the reactivity of acyl derivatives is leaving group ability, which is related to acidity. Weak bases are better leaving groups than strong bases; a species with a strong conjugate acid (e.g. hydrochloric acid) will be a better leaving group than a species with a weak conjugate acid (e.g. acetic acid). Thus, chloride ion is a better leaving group than acetate ion. The reactivity of acyl compounds towards nucleophiles decreases as the basicity of the leaving group increases, as the table shows.[4]
| Compound Name | Structure | Leaving Group | pKa of Conjugate Acid |
|---|---|---|---|
| Acetyl chloride | 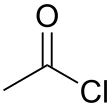 |
−7 | |
| Acetic anhydride | 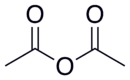 |
 |
4.76 |
| Ethyl acetate |  |
15.9 | |
| Acetamide | 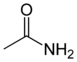 |
38 | |
| Acetate anion | |
N/a | N/a |

Another factor that plays a role in determining the reactivity of acyl compounds is resonance. Amides exhibit two main resonance forms. Both are major contributors to the overall structure, so much so that the amide bond between the carbonyl carbon and the amide nitrogen has significant double bond character. The energy barrier for rotation about an amide bond is 75–85 kJ/mol (18–20 kcal/mol), much larger than values observed for normal single bonds. For example, the C–C bond in ethane has an energy barrier of only 12 kJ/mol (3 kcal/mol).[3] Once a nucleophile attacks and a tetrahedral intermediate is formed, the energetically favorable resonance effect is lost. This helps explain why amides are one of the least reactive acyl derivatives.[4]
Esters exhibit less resonance stabilization than amides, so the formation of a tetrahedral intermediate and subsequent loss of resonance is not as energetically unfavorable. Anhydrides experience even weaker resonance stabilization, since the resonance is split between two carbonyl groups, and are more reactive than esters and amides. In acid halides, there is very little resonance, so the energetic penalty for forming a tetrahedral intermediate is small. This helps explain why acid halides are the most reactive acyl derivatives.[4]
Reactions of acyl derivatives
Many nucleophilic acyl substitution reactions involve converting one acyl derivative into another. In general, conversions between acyl derivatives must proceed from a relatively reactive compound to a less reactive one to be practical; an acid chloride can easily be converted to an ester, but converting an ester directly to an acid chloride is essentially impossible. When converting between acyl derivatives, the product will always be more stable than the starting compound.
Nucleophilic acyl substitution reactions that do not involve interconversion between acyl derivatives are also possible. For example, amides and carboxylic acids react with Grignard reagents to produce ketones. An overview of the reactions that each type of acyl derivative can participate in is presented here.
Acid halides
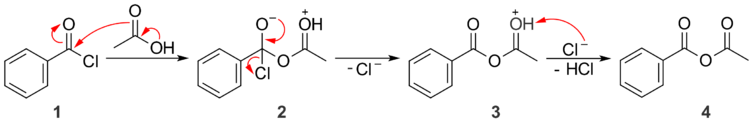
Acid halides are the most reactive acyl derivatives, and can easily be converted into any of the others. Acid halides will react with carboxylic acids to form anhydrides. If the structure of the acid and the acid chloride are different, the product is a mixed anhydride. First, the carboxylic acid attacks the acid chloride (1) to give tetrahedral intermediate 2. The tetrahedral intermediate collapses, ejecting chloride ion as the leaving group and forming oxonium species 3. Deprotonation gives the mixed anhydride, 4, and an equivalent of HCl.
Alcohols and amines react with acid halides to produce esters and amides, respectively, in a reaction formally known as the Schotten-Baumann reaction.[5] Acid halides hydrolyze in the presence of water to produce carboxylic acids, but this type of reaction is rarely useful, since carboxylic acids are typically used to synthesize acid halides. Most reactions with acid halides are carried out in the presence of a non-nucleophilic base, such as pyridine, to neutralize the hydrohalic acid that is formed as a byproduct.
Acid halides will react with carbon nucleophiles, such as Grignards and enolates, though mixtures of products can result. While a carbon nucleophile will react with the acid halide first to produce a ketone, the ketone is also susceptible to nucleophilic attack, and can be converted to a tertiary alcohol. For example, when benzoyl chloride (1) is treated with two equivalents of a Grignard reagent, such as methyl magnesium bromide (MeMgBr), 2-phenyl-2-propanol (3) is obtained in excellent yield. Although acetophenone (2) is an intermediate in this reaction, it is impossible to isolate because it reacts with a second equivalent of MeMgBr rapidly after being formed.[6]

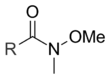
Unlike most other carbon nucleophiles, lithium dialkylcuprates – often called Gilman reagents – can add to acid halides just once to give ketones. The reaction between an acid halide and a Gilman reagent is not a nucleophilic acyl substitution reaction, however, and is thought to proceed via a radical pathway.[2] The Weinreb ketone synthesis can also be used to convert acid halides to ketones. In this reaction, the acid halide is first converted to an N–methoxy–N–methylamide, known as a Weinreb amide. When a carbon nucleophile – such as a Grignard or organolithium reagent – adds to a Weinreb amide, the metal is chelated by the carbonyl and N–methoxy oxygens, preventing further nucleophilic additions.[7]
In the Friedel–Crafts acylation, acid halides act as electrophiles for electrophilic aromatic substitution. A Lewis acid – such as zinc chloride (ZnCl2), iron(III) chloride (FeCl3), or aluminum chloride (AlCl3) – coordinates to the halogen on the acid halide, activating the compound towards nucleophilic attack by an activated aromatic ring. For especially electron-rich aromatic rings, the reaction will proceed without a Lewis acid.[8]
Thioesters
The chemistry of thioesters and acid halides is similar, the reactivity being reminiscent of, but milder, than acid chlorides.
Anhydrides
The chemistry of acid halides and anhydrides is similar. While anhydrides cannot be converted to acid halides, they can be converted to the remaining acyl derivatives. Anhydrides also participate in Schotten–Baumann-type reactions to furnish esters and amides from alcohols and amines, and water can hydrolyze anhydrides to their corresponding acids. As with acid halides, anhydrides can also react with carbon nucleophiles to furnish ketones and/or tertiary alcohols, and can participate in both the Friedel–Crafts acylation and the Weinreb ketone synthesis.[8] Unlike acid halides, however, anhydrides do not react with Gilman reagents.[2]
The reactivity of anhydrides can be increased by using a catalytic amount of N,N-dimethylaminopyridine, or DMAP. Pyridine can also be used for this purpose, and acts via a similar mechanism.[5]
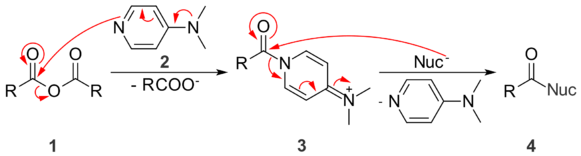
First, DMAP (2) attacks the anhydride (1) to form a tetrahedral intermediate, which collapses to eliminate a carboxylate ion to give amide 3. This intermediate amide is more activated towards nucleophilic attack than the original anhydride, because dimethylaminopyridine is a better leaving group than a carboxylate. In the final set of steps, a nucleophile (Nuc) attacks 3 to give another tetrahedral intermediate. When this intermediate collapses to give the product 4, the pyridine group is eliminated and its aromaticity is restored – a powerful driving force, and the reason why the pyridine compound is a better leaving group than a carboxylate ion.
Esters
Esters are less reactive than acid halides and anhydrides. As with more reactive acyl derivatives, they can react with ammonia and primary and secondary amines to give amides, though this type of reaction is not often used, since acid halides give better yields. Esters can be converted to other esters in a process known as transesterification. Transesterification can be either acid- or base-catalyzed, and involves the reaction of an ester with an alcohol. Unfortunately, because the leaving group is also an alcohol, the forward and reverse reactions will often occur at similar rates. Using a large excess of the reactant alcohol or removing the leaving group alcohol (e.g. via distillation) will drive the forward reaction towards completion, in accordance with Le Chatelier's principle.[9]
Acid-catalyzed hydrolysis of esters is also an equilibrium process – essentially the reverse of the Fischer esterification reaction. Because an alcohol (which acts as the leaving group) and water (which acts as the nucleophile) have similar pKa values, the forward and reverse reactions compete with each other. As in transesterification, using a large excess of reactant (water) or removing one of the products (the alcohol) can promote the forward reaction.
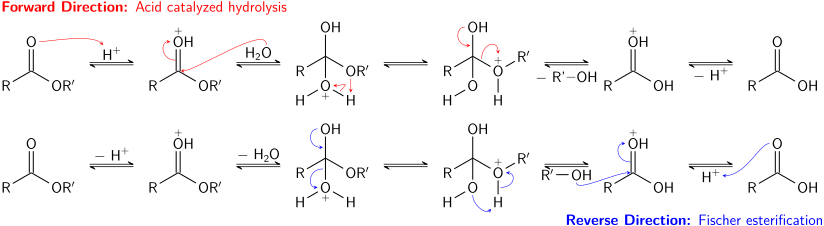
Basic hydrolysis of esters, known as saponification, is not an equilibrium process; a full equivalent of base is consumed in the reaction, which produces one equivalent of alcohol and one equivalent of a carboxylate salt. The saponification of esters of fatty acids is an industrially important process, used in the production of soap.[9]
Esters can undergo a variety of reactions with carbon nucleophiles. As with acid halides and anhyrides, they will react with an excess of a Grignard reagent to give tertiary alcohols. Esters also react readily with enolates. In the Claisen condensation, an enolate of one ester (1) will attack the carbonyl group of another ester (2) to give tetrahedral intermediate 3. The intermediate collapses, forcing out an alkoxide (R'O−) and producing β-keto ester 4.
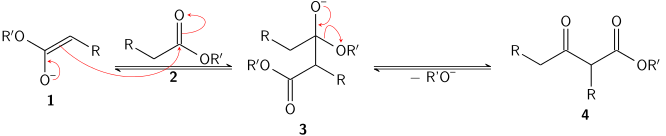
Crossed Claisen condensations, in which the enolate and nucleophile are different esters, are also possible. An intramolecular Claisen condensation is called a Dieckmann condensation or Dieckmann cyclization, since it can be used to form rings. Esters can also undergo condensations with ketone and aldehyde enolates to give β-dicarbonyl compounds.[10] A specific example of this is the Baker–Venkataraman rearrangement, in which an aromatic ortho-acyloxy ketone undergoes an intramolecular nucleophilic acyl substitution and subsequent rearrangement to form an aromatic β-diketone.[11] The Chan rearrangement is another example of a rearrangement resulting from an intramolecular nucleophilic acyl substitution reaction.
Amides
Because of their low reactivity, amides do not participate in nearly as many nucleophilic substitution reactions as other acyl derivatives do. Amides are stable to water, and are roughly 100 times more stable towards hydrolysis than esters.[3] Amides can, however, be hydrolyzed to carboxylic acids in the presence of acid or base. The stability of amide bonds has biological implications, since the amino acids that make up proteins are linked with amide bonds. Amide bonds are resistant enough to hydrolysis to maintain protein structure in aqueous environments, but are susceptible enough that they can be broken when necessary.[3]
Primary and secondary amides do not react favorably with carbon nucleophiles. Grignard reagents and organolithiums will act as bases rather than nucleophiles, and will simply deprotonate the amide. Tertiary amides do not experience this problem, and react with carbon nucleophiles to give ketones; the amide anion (NR2−) is a very strong base and thus a very poor leaving group, so nucleophilic attack only occurs once. When reacted with carbon nucleophiles, N,N-dimethylformamide (DMF) can be used to introduce a formyl group.[12]
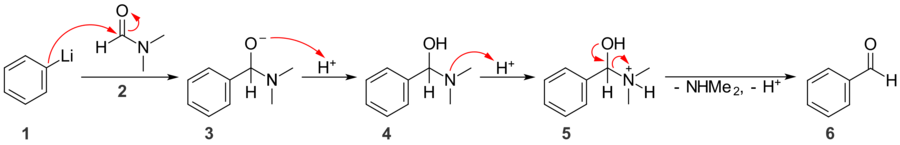
Here, phenyllithium 1 attacks the carbonyl group of DMF 2, giving tetrahedral intermediate 3. Because the dimethylamide anion is a poor leaving group, the intermediate does not collapse and another nucleophilic addition does not occur. Upon acidic workup, the alkoxide is protonated to give 4, then the amine is protonated to give 5. Elimination of a neutral molecule of dimethylamine and loss of a proton give benzaldehyde, 6.
Carboxylic acids
Carboxylic acids are not especially reactive towards nucleophilic substitution, though they can be converted to other acyl derivatives. Converting a carboxylic acid to an amide is possible, but not straightforward. Instead of acting as a nucleophile, an amine will react as a base in the presence of a carboxylic acid to give the ammonium carboxylate salt. Heating the salt to above 100 °C will drive off water and lead to the formation of the amide. This method of synthesizing amides is industrially important, and has laboratory applications as well.[13] In the presence of a strong acid catalyst, carboxylic acids can condense to form acid anhydrides. The condensation produces water, however, which can hydrolyze the anhydride back to the starting carboxylic acids. Thus, the formation of the anhydride via condensation is an equilibrium process.
Under acid-catalyzed conditions, carboxylic acids will react with alcohols to form esters via the Fischer esterification reaction, which is also an equilibrium process. Alternatively, diazomethane can be used to convert an acid to an ester. While esterification reactions with diazomethane often give quantitative yields, diazomethane is only useful for forming methyl esters.[13]
Thionyl chloride can be used to convert carboxylic acids to their corresponding acyl chlorides. First, carboxylic acid 1 attacks thionyl chloride, and chloride ion leaves. The resulting oxonium ion 2 is activated towards nucleophilic attack and has a good leaving group, setting it apart from a normal carboxylic acid. In the next step, 2 is attacked by chloride ion to give tetrahedral intermediate 3, a chlorosulfite. The tetrahedral intermediate collapses with the loss of sulfur dioxide and chloride ion, giving protonated acyl chloride 4. Chloride ion can remove the proton on the carbonyl group, giving the acyl chloride 5 with a loss of HCl.

Phosphorus(III) chloride (PCl3) and phosphorus(V) chloride (PCl5) will also convert carboxylic acids to acid chlorides, by a similar mechanism. One equivalent of PCl3 can react with three equivalents of acid, producing one equivalent of H3PO3, or phosphorus acid, in addition to the desired acid chloride. PCl5 reacts with carboxylic acids in a 1:1 ratio, and produces phosphorus(V) oxychloride (POCl3) and hydrogen chloride (HCl) as byproducts.
Carboxylic acids react with Grignard reagents and organolithiums to form ketones. The first equivalent of nucleophile acts as a base and deprotonates the acid. A second equivalent will attack the carbonyl group to create a geminal alkoxide dianion, which is protonated upon workup to give the hydrate of a ketone. Because most ketone hydrates are unstable relative to their corresponding ketones, the equilibrium between the two is shifted heavily in favor of the ketone. For example, the equilibrium constant for the formation of acetone hydrate from acetone is only 0.002. The carboxylic group is the most acidic in organic compounds.[14]
See also
- Nucleophilic aliphatic substitution
- Nucleophilic aromatic substitution
- Nucleophilic abstraction
References
- Wade 2010, pp. 996–997.
- McMurry, John (1996). Organic Chemistry (4th ed.). Pacific Grove, CA: Brooks/Cole Publishing Company. pp. 820–821. ISBN 0534238327.
- Carey, Francis A. (2006). Organic Chemistry (6th ed.). New York: McGraw-Hill. pp. 866–868. ISBN 0072828374.
- Wade 2010, pp. 998–999.
- Kürti, László; Barbara Czakó (2005). Strategic Applications of Named Reactions in Organic Synthesis. London: Elsevier Academic Press. p. 398. ISBN 0124297854.
- McMurry 1996, pp. 826–827.
- Kürti and Czakó 2005, p. 478.
- Kürti and Czakó 2005, p. 176.
- Wade 2010, pp. 1005–1009.
- Carey 2006, pp. 919–924.
- Kürti and Czakó 2005, p. 30.
- Alan R. Katritzky; Meth-Cohn, Otto; Charles Rees, eds. (1995). Comprehensive Organic Functional Group Transformations. 3 (1st ed.). Oxford: Pergamon Press. p. 90. ISBN 0080423248.
- Wade 2010, pp. 964–965.
- Wade 2010, p. 838.
External links
- Reaction of acetic anhydride with acetone in Organic Syntheses Coll. Vol. 3, p. 16; Vol. 20, p. 6 Article