Monoclonal antibody
Monoclonal antibodies (mAb or moAb) are antibodies that are made by identical immune cells which are all clones belonging to a unique parent cell. Monoclonal antibodies can have monovalent affinity, in that they bind to the same epitope (the part of an antigen that is recognized by the antibody). In contrast, polyclonal antibodies bind to multiple epitopes and are usually made by several different plasma cell (antibody secreting immune cell) lineages. Bispecific monoclonal antibodies can also be engineered, by increasing the therapeutic targets of one single monoclonal antibody to two epitopes.
Given almost any substance, it is possible to produce monoclonal antibodies that specifically bind to that substance; they can then serve to detect or purify that substance. This has become an important tool in biochemistry, molecular biology, and medicine. When used as medications, non-proprietary drug names end in -mab (see "Nomenclature of monoclonal antibodies") and many immunotherapy specialists use the word mab anacronymically.
History
The idea of "magic bullets" was first proposed by Paul Ehrlich, who, at the beginning of the 20th century, postulated that, if a compound could be made that selectively targeted a disease-causing organism, then a toxin for that organism could be delivered along with the agent of selectivity. He and Élie Metchnikoff received the 1908 Nobel Prize for Physiology or Medicine for this work.
In the 1970s, the B-cell cancer multiple myeloma was known. It was understood that these cancerous B-cells all produce a single type of antibody (a paraprotein). This was used to study the structure of antibodies, but it was not yet possible to produce identical antibodies specific to a given antigen.[3]:324
Production of monoclonal antibodies involving human–mouse hybrid cells was first described by Jerrold Schwaber in 1973[4] and remains widely cited among those using human-derived hybridomas.[5]
In 1975, Georges Köhler and César Milstein succeeded in making fusions of myeloma cell lines with B cells to create hybridomas that could produce antibodies, specific to known antigens and that were immortalized.[6] They and Niels Kaj Jerne shared the Nobel Prize in Physiology or Medicine in 1984 for the discovery.[6]
In 1988, Greg Winter and his team pioneered the techniques to humanize monoclonal antibodies,[7] eliminating the reactions that many monoclonal antibodies caused in some patients.
In 2018, James P. Allison and Tasuku Honjo received the Nobel Prize in Physiology or Medicine for their discovery of cancer therapy by inhibition of negative immune regulation, using monoclonal antibodies that prevent inhibitory linkages.[8]
Production




Hybridoma development
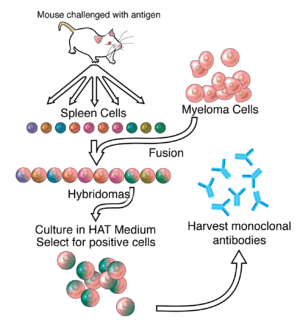
Much of the work behind production of monoclonal antibodies is rooted in the production of hybridomas, which involves identifying antigen-specific plasma/plasmablast cells (ASPCs) that produce antibodies specific to an antigen of interest and fusing these cells with myeloma cells. Rabbit B-cells can be used to form a rabbit hybridoma. Polyethylene glycol is used to fuse adjacent plasma membranes,[9] but the success rate is low, so a selective medium in which only fused cells can grow is used. This is possible because myeloma cells have lost the ability to synthesize hypoxanthine-guanine-phosphoribosyl transferase (HGPRT), an enzyme necessary for the salvage synthesis of nucleic acids. The absence of HGPRT is not a problem for these cells unless the de novo purine synthesis pathway is also disrupted. Exposing cells to aminopterin (a folic acid analogue, which inhibits dihydrofolate reductase, DHFR), makes them unable to use the de novo pathway and become fully auxotrophic for nucleic acids, thus requiring supplementation to survive.
The selective culture medium is called HAT medium because it contains hypoxanthine, aminopterin and thymidine. This medium is selective for fused (hybridoma) cells. Unfused myeloma cells cannot grow because they lack HGPRT and thus cannot replicate their DNA. Unfused spleen cells cannot grow indefinitely because of their limited life span. Only fused hybrid cells referred to as hybridomas, are able to grow indefinitely in the medium because the spleen cell partner supplies HGPRT and the myeloma partner has traits that make it immortal (similar to a cancer cell).
This mixture of cells is then diluted and clones are grown from single parent cells on microtitre wells. The antibodies secreted by the different clones are then assayed for their ability to bind to the antigen (with a test such as ELISA or Antigen Microarray Assay) or immuno-dot blot. The most productive and stable clone is then selected for future use.
The hybridomas can be grown indefinitely in a suitable cell culture medium. They can also be injected into mice (in the peritoneal cavity, surrounding the gut). There, they produce tumors secreting an antibody-rich fluid called ascites fluid.
The medium must be enriched during in vitro selection to further favour hybridoma growth. This can be achieved by the use of a layer of feeder fibrocyte cells or supplement medium such as briclone. Culture-media conditioned by macrophages can be used. Production in cell culture is usually preferred as the ascites technique is painful to the animal. Where alternate techniques exist, ascites is considered unethical.[10]
Novel mAb development technology
Several monoclonal antibody technologies had been developed recently,[11] such as phage display,[12] single B cell culture,[13] single cell amplification from various B cell populations[14][15][16][17][18] and single plasma cell interrogation technologies. Different from traditional hybridoma technology, the newer technologies use molecular biology techniques to amplify the heavy and light chains of the antibody genes by PCR and produce in either bacterial or mammalian systems with recombinant technology. One of the advantages of the new technologies is applicable to multiple animals, such as rabbit, llama, chicken and other common experimental animals in the laboratory.
Purification
After obtaining either a media sample of cultured hybridomas or a sample of ascites fluid, the desired antibodies must be extracted. Cell culture sample contaminants consist primarily of media components such as growth factors, hormones and transferrins. In contrast, the in vivo sample is likely to have host antibodies, proteases, nucleases, nucleic acids and viruses. In both cases, other secretions by the hybridomas such as cytokines may be present. There may also be bacterial contamination and, as a result, endotoxins that are secreted by the bacteria. Depending on the complexity of the media required in cell culture and thus the contaminants, one or the other method (in vivo or in vitro) may be preferable.
The sample is first conditioned, or prepared for purification. Cells, cell debris, lipids, and clotted material are first removed, typically by centrifugation followed by filtration with a 0.45 µm filter. These large particles can cause a phenomenon called membrane fouling in later purification steps. In addition, the concentration of product in the sample may not be sufficient, especially in cases where the desired antibody is produced by a low-secreting cell line. The sample is therefore concentrated by ultrafiltration or dialysis.
Most of the charged impurities are usually anions such as nucleic acids and endotoxins. These can be separated by ion exchange chromatography.[19] Either cation exchange chromatography is used at a low enough pH that the desired antibody binds to the column while anions flow through, or anion exchange chromatography is used at a high enough pH that the desired antibody flows through the column while anions bind to it. Various proteins can also be separated along with the anions based on their isoelectric point (pI). In proteins, the isoelectric point (pI) is defined as the pH at which a protein has no net charge. When the pH > pI, a protein has a net negative charge, and when the pH < pI, a protein has a net positive charge. For example, albumin has a pI of 4.8, which is significantly lower than that of most monoclonal antibodies, which have a pI of 6.1. Thus, at a pH between 4.8 and 6.1, the average charge of albumin molecules is likely to be more negative, while mAbs molecules are positively charged and hence it is possible to separate them. Transferrin, on the other hand, has a pI of 5.9, so it cannot be easily separated by this method. A difference in pI of at least 1 is necessary for a good separation.
Transferrin can instead be removed by size exclusion chromatography. This method is one of the more reliable chromatography techniques. Since we are dealing with proteins, properties such as charge and affinity are not consistent and vary with pH as molecules are protonated and deprotonated, while size stays relatively constant. Nonetheless, it has drawbacks such as low resolution, low capacity and low elution times.
A much quicker, single-step method of separation is protein A/G affinity chromatography. The antibody selectively binds to protein A/G, so a high level of purity (generally >80%) is obtained. However, this method may be problematic for antibodies that are easily damaged, as harsh conditions are generally used. A low pH can break the bonds to remove the antibody from the column. In addition to possibly affecting the product, low pH can cause protein A/G itself to leak off the column and appear in the eluted sample. Gentle elution buffer systems that employ high salt concentrations are available to avoid exposing sensitive antibodies to low pH. Cost is also an important consideration with this method because immobilized protein A/G is a more expensive resin.
To achieve maximum purity in a single step, affinity purification can be performed, using the antigen to provide specificity for the antibody. In this method, the antigen used to generate the antibody is covalently attached to an agarose support. If the antigen is a peptide, it is commonly synthesized with a terminal cysteine, which allows selective attachment to a carrier protein, such as KLH during development and to support purification. The antibody-containing medium is then incubated with the immobilized antigen, either in batch or as the antibody is passed through a column, where it selectively binds and can be retained while impurities are washed away. An elution with a low pH buffer or a more gentle, high salt elution buffer is then used to recover purified antibody from the support.
Antibody heterogeneity
Product heterogeneity is common in monoclonal antibodies and other recombinant biological products and is typically introduced either upstream during expression or downstream during manufacturing.
These variants are typically aggregates, deamidation products, glycosylation variants, oxidized amino acid side chains, as well as amino and carboxyl terminal amino acid additions.[20] These seemingly minute structural changes can affect preclinical stability and process optimization as well as therapeutic product potency, bioavailability and immunogenicity. The generally accepted purification method of process streams for monoclonal antibodies includes capture of the product target with protein A, elution, acidification to inactivate potential mammalian viruses, followed by ion chromatography, first with anion beads and then with cation beads.
Displacement chromatography has been used to identify and characterize these often unseen variants in quantities that are suitable for subsequent preclinical evaluation regimens such as animal pharmacokinetic studies.[21][22] Knowledge gained during the preclinical development phase is critical for enhanced product quality understanding and provides a basis for risk management and increased regulatory flexibility. The recent Food and Drug Administration's Quality by Design initiative attempts to provide guidance on development and to facilitate design of products and processes that maximizes efficacy and safety profile while enhancing product manufacturability.[23]
Recombinant
The production of recombinant monoclonal antibodies involves repertoire cloning, CRISPR/Cas9, or phage display/yeast display technologies.[24] Recombinant antibody engineering involves antibody production by the use of viruses or yeast, rather than mice. These techniques rely on rapid cloning of immunoglobulin gene segments to create libraries of antibodies with slightly different amino acid sequences from which antibodies with desired specificities can be selected.[25] The phage antibody libraries are a variant of phage antigen libraries.[26] These techniques can be used to enhance the specificity with which antibodies recognize antigens, their stability in various environmental conditions, their therapeutic efficacy and their detectability in diagnostic applications.[27] Fermentation chambers have been used for large scale antibody production.
Chimeric antibodies
While mouse and human antibodies are structurally similar, the differences between them were sufficient to invoke an immune response when murine monoclonal antibodies were injected into humans, resulting in their rapid removal from the blood, as well as systemic inflammatory effects and the production of human anti-mouse antibodies (HAMA).
Recombinant DNA has been explored since the late 1980s to increase residence times. In one approach, mouse DNA encoding the binding portion of a monoclonal antibody was merged with human antibody-producing DNA in living cells. The expression of this "chimeric" or "humanised" DNA through cell culture yielded part-mouse, part-human antibodies.[28][29]
Human antibodies
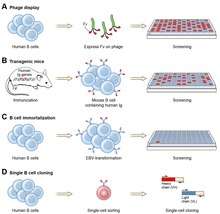
Ever since the discovery that monoclonal antibodies could be generated, scientists have targeted the creation of fully human products to reduce the side effects of humanised or chimeric antibodies. Several successful approaches have been identified: transgenic mice,[30] phage display[12] and single B cell cloning:[11]
As of November 2016, thirteen of the nineteen fully human monoclonal antibody therapeutics on the market were derived from transgenic mice technology.
Adopting organizations who market transgenic technology include:
- Medarex — which marketed the UltiMab platform. Medarex was acquired in July 2009 by Bristol Myers Squibb[31]
- Abgenix — which marketed the Xenomouse technology. Abgenix was acquired in April 2006 by Amgen.[32]
- Regeneron Pharmaceuticals VelocImmune technology.[33]
- Kymab - who market their Kymouse technology.[34]
- Open Monoclonal Technology's OmniRat™ and OmniMouse™ platform.[35]
- TRIANNI, Inc. – who market their TRIANNI Mouse platform.[36]
- Ablexis, LLC - who market their AlivaMab Mouse platform.[37]
Phage display can be used to express variable antibody domains on filamentous phage coat proteins (Phage major coat protein).[38][39][40] These phage display antibodies can be used for various research applications.[41][42] ProAb was announced in December 1997[43] and involved high throughput screening of antibody libraries against diseased and non-diseased tissue, whilst Proximol used a free radical enzymatic reaction to label molecules in proximity to a given protein.[44][45]
Monoclonal antibodies have been approved to treat cancer, cardiovascular disease, inflammatory diseases, macular degeneration, transplant rejection, multiple sclerosis and viral infection.
In August 2006, the Pharmaceutical Research and Manufacturers of America reported that U.S. companies had 160 different monoclonal antibodies in clinical trials or awaiting approval by the Food and Drug Administration.[46]
Cost
With respect to price, MAbs are unusual like vinyl records, or digital media: once you've made the first one - the first music (or program or other data) and a master - or the first mAb - it's almost free, in comparison, to make more copies. They are priced to make profit and (if not a biosimilar) recoup the typically large investment costs, and where there are no price controls, such as the United States, prices are set very high if they provide great value. Seven University of Pittsburgh researchers concluded, "The annual price of mAb therapies is about $100,000 higher in oncology and hematology than in other disease states," comparing them on a per patient basis, to those for cardiovascular or metabolic disorders, immunology, infectious diseases, allergy, and ophthalmology.[47]
Applications
Diagnostic tests
Once monoclonal antibodies for a given substance have been produced, they can be used to detect the presence of this substance. Proteins can be detected using the Western blot and immuno dot blot tests. In immunohistochemistry, monoclonal antibodies can be used to detect antigens in fixed tissue sections, and similarly, immunofluorescence can be used to detect a substance in either frozen tissue section or live cells.
Analytic and chemical uses
Antibodies can also be used to purify their target compounds from mixtures, using the method of immunoprecipitation.
Therapeutic uses
Therapeutic monoclonal antibodies act through multiple mechanisms, such as blocking of targeted molecule functions, inducing apoptosis in cells which express the target, or by modulating signalling pathways.[48][49]
Cancer treatment
One possible treatment for cancer involves monoclonal antibodies that bind only to cancer-cell-specific antigens and induce an immune response against the target cancer cell. Such mAbs can be modified for delivery of a toxin, radioisotope, cytokine or other active conjugate or to design bispecific antibodies that can bind with their Fab regions both to target antigen and to a conjugate or effector cell. Every intact antibody can bind to cell receptors or other proteins with its Fc region.
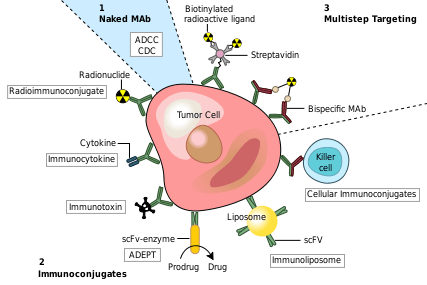
MAbs approved by the FDA for cancer include:[51]
Autoimmune diseases
Monoclonal antibodies used for autoimmune diseases include infliximab and adalimumab, which are effective in rheumatoid arthritis, Crohn's disease, ulcerative colitis and ankylosing spondylitis by their ability to bind to and inhibit TNF-α.[52] Basiliximab and daclizumab inhibit IL-2 on activated T cells and thereby help prevent acute rejection of kidney transplants.[52] Omalizumab inhibits human immunoglobulin E (IgE) and is useful in treating moderate-to-severe allergic asthma.
Examples of therapeutic monoclonal antibodies
Monoclonal antibodies for research applications can be found directly from antibody suppliers, or through use of a specialist search engine like CiteAb. Below are examples of clinically important monoclonal antibodies.
| Main category | Type | Application | Mechanism/Target | Mode |
|---|---|---|---|---|
| Anti- inflammatory | infliximab[52] | inhibits TNF-α | chimeric | |
| adalimumab | inhibits TNF-α | human | ||
| basiliximab[52] |
|
inhibits IL-2 on activated T cells | chimeric | |
| daclizumab[52] |
|
inhibits IL-2 on activated T cells | humanized | |
| omalizumab |
|
inhibits human immunoglobulin E (IgE) | humanized | |
| Anti-cancer | gemtuzumab[52] |
|
targets myeloid cell surface antigen CD33 on leukemia cells | humanized |
| alemtuzumab[52] |
|
targets an antigen CD52 on T- and B-lymphocytes | humanized | |
| rituximab[52] |
|
targets phosphoprotein CD20 on B lymphocytes | chimeric | |
| trastuzumab |
|
targets the HER2/neu (erbB2) receptor | humanized | |
| nimotuzumab |
|
EGFR inhibitor | humanized | |
| cetuximab |
|
EGFR inhibitor | chimeric | |
| bevacizumab & ranibizumab |
|
inhibits VEGF | humanized | |
| Anti-cancer and anti-viral | bavituximab[53] |
|
immunotherapy, targets phosphatidylserine[53] | chimeric |
| Other | palivizumab[52] |
|
inhibits an RSV fusion (F) protein | humanized |
| abciximab[52] |
|
inhibits the receptor GpIIb/IIIa on platelets | chimeric | |
Side effects
Several monoclonal antibodies, such as Bevacizumab and Cetuximab, can cause different kinds of side effects.[54] These side effects can be categorized into common and serious side effects.[55]
Some common side effects include:
Among the possible serious side effects are:
- Anaphylaxis
- Bleeding
- Arterial and venous blood clots
- Autoimmune thyroiditis
- Hypothyroidism
- Hepatitis
- Heart failure
- Cancer
- Anemia
- Decrease in white blood cells
- Stomatitis
- Enterocolitis
- Gastrointestinal perforation
- Mucositis[56]
See also
- Affimer
- Antibody mimetic
- Aptamer
- Immunotoxins, which sometimes use monoclonal antibodies as the targeting mechanism
- List of monoclonal antibodies
- Monoclonal antibody therapy
- Nomenclature of monoclonal antibodies
- Polyclonal antibodies
- Monoclonal Antibody Journal
References
- "Cytochrome P450 Mediated Drug and Carcinogen Metabolism using Monoclonal Antibodies". home.ccr.cancer.gov. Retrieved 2018-04-02.
- Gelboin HV, Krausz KW, Gonzalez FJ, Yang TJ (November 1999). "Inhibitory monoclonal antibodies to human cytochrome P450 enzymes: a new avenue for drug discovery". Trends in Pharmacological Sciences. 20 (11): 432–8. doi:10.1016/S0165-6147(99)01382-6. PMID 10542439.
- Tansey EM, Catterall PP (July 1994). "Monoclonal antibodies: a witness seminar in contemporary medical history". Medical History. 38 (3): 322–7. doi:10.1017/s0025727300036632. PMC 1036884. PMID 7934322.
- Schwaber J, Cohen EP (August 1973). "Human x mouse somatic cell hybrid clone secreting immunoglobulins of both parental types". Nature. 244 (5416): 444–7. doi:10.1038/244444a0. PMID 4200460.
- Cambrosio A, Keating P (1992). "Between fact and technique: the beginnings of hybridoma technology". Journal of the History of Biology. 25 (2): 175–230. doi:10.1007/BF00162840. PMID 11623041.
- Marks LV, A Healthcare Revolution in the Making: The Story of César Milstein and Monoclonal Antibodies: Making monoclonal antibodies
- Riechmann L, Clark M, Waldmann H, Winter G (March 1988). "Reshaping human antibodies for therapy". Nature. 332 (6162): 323–7. Bibcode:1988Natur.332..323R. doi:10.1038/332323a0. PMID 3127726.
- Altmann DM (November 2018). "A Nobel Prize-worthy pursuit: cancer immunology and harnessing immunity to tumour neoantigens". Immunology. 155 (3): 283–284. doi:10.1111/imm.13008. PMC 6187215. PMID 30320408.
- Yang J1, Shen MH. Polyethylene glycol-mediated cell fusion. Methods Mol Biol. 2006; 325:59-66.
- National Research Council (US) Committee on Methods of Producing Monoclonal Antibodies. Recommendation 1: Executive Summary: Monoclonal Antibody Production. Washington (DC): National Academies Press (US); 1999. ISBN 978-0-309-07511-4
- Ho M (June 2018). "Inaugural Editorial: Searching for Magic Bullets". Antibody Therapeutics. 1 (1): 1–5. doi:10.1093/abt/tby001. PMC 6086361. PMID 30101214.
- Ho M, Feng M, Fisher RJ, Rader C, Pastan I (May 2011). "A novel high-affinity human monoclonal antibody to mesothelin". International Journal of Cancer. 128 (9): 2020–30. doi:10.1002/ijc.25557. PMC 2978266. PMID 20635390.
- Seeber S, Ros F, Thorey I, Tiefenthaler G, Kaluza K, Lifke V, et al. (2014). "A robust high throughput platform to generate functional recombinant monoclonal antibodies using rabbit B cells from peripheral blood". PLOS ONE. 9 (2): e86184. Bibcode:2014PLoSO...986184S. doi:10.1371/journal.pone.0086184. PMC 3913575. PMID 24503933.
- Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC (September 2003). "Predominant autoantibody production by early human B cell precursors". Science. 301 (5638): 1374–7. Bibcode:2003Sci...301.1374W. doi:10.1126/science.1086907. PMID 12920303.
- Koelsch K, Zheng NY, Zhang Q, Duty A, Helms C, Mathias MD, et al. (June 2007). "Mature B cells class switched to IgD are autoreactive in healthy individuals". The Journal of Clinical Investigation. 117 (6): 1558–65. doi:10.1172/JCI27628. PMC 1866247. PMID 17510706.
- Smith K, Garman L, Wrammert J, Zheng NY, Capra JD, Ahmed R, Wilson PC (2009-01-01). "Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen". Nature Protocols. 4 (3): 372–84. doi:10.1038/nprot.2009.3. PMC 2750034. PMID 19247287.
- Duty JA, Szodoray P, Zheng NY, Koelsch KA, Zhang Q, Swiatkowski M, et al. (January 2009). "Functional anergy in a subpopulation of naive B cells from healthy humans that express autoreactive immunoglobulin receptors". The Journal of Experimental Medicine. 206 (1): 139–51. doi:10.1084/jem.20080611. PMC 2626668. PMID 19103878.
- Huang J, Doria-Rose NA, Longo NS, Laub L, Lin CL, Turk E, et al. (October 2013). "Isolation of human monoclonal antibodies from peripheral blood B cells". Nature Protocols. 8 (10): 1907–15. doi:10.1038/nprot.2013.117. PMC 4844175. PMID 24030440.
- Vlasak J, Ionescu R (December 2008). "Heterogeneity of monoclonal antibodies revealed by charge-sensitive methods". Current Pharmaceutical Biotechnology. 9 (6): 468–81. doi:10.2174/138920108786786402. PMID 19075686.
- Beck A, Wurch T, Bailly C, Corvaia N (May 2010). "Strategies and challenges for the next generation of therapeutic antibodies". Nature Reviews. Immunology. 10 (5): 345–52. doi:10.1038/nri2747. PMID 20414207.
- Khawli LA, Goswami S, Hutchinson R, Kwong ZW, Yang J, Wang X, et al. (2010). "Charge variants in IgG1: Isolation, characterization, in vitro binding properties and pharmacokinetics in rats". mAbs. 2 (6): 613–24. doi:10.4161/mabs.2.6.13333. PMC 3011216. PMID 20818176.
- Zhang T, Bourret J, Cano T (August 2011). "Isolation and characterization of therapeutic antibody charge variants using cation exchange displacement chromatography". Journal of Chromatography. A. 1218 (31): 5079–86. doi:10.1016/j.chroma.2011.05.061. PMID 21700290.
- Rathore AS, Winkle H (January 2009). "Quality by design for biopharmaceuticals". Nature Biotechnology. 27 (1): 26–34. doi:10.1038/nbt0109-26. PMID 19131992.
- van der Schoot JM, Fennemann FL, Valente M, Dolen Y, Hagemans IM, Becker AM, et al. (August 2019). "Functional diversification of hybridoma-produced antibodies by CRISPR/HDR genomic engineering". Science Advances. 5 (8): eaaw1822. doi:10.1126/sciadv.aaw1822. PMC 6713500. PMID 31489367.
- Siegel DL (January 2002). "Recombinant monoclonal antibody technology". Transfusion Clinique et Biologique. 9 (1): 15–22. doi:10.1016/S1246-7820(01)00210-5. PMID 11889896.
- "Dr. George Pieczenik". LMB Alumni. MRC Laboratory of Molecular Biology (LMB). 17 September 2009. Archived from the original on 23 December 2012. Retrieved 17 November 2012.
- Schmitz U, Versmold A, Kaufmann P, Frank HG (2000). "Phage display: a molecular tool for the generation of antibodies--a review". Placenta. 21 Suppl A (Suppl A): S106-12. doi:10.1053/plac.1999.0511. PMID 10831134.
- Boulianne GL, Hozumi N, Shulman MJ (1984). "Production of functional chimaeric mouse/human antibody". Nature. 312 (5995): 643–6. doi:10.1038/312643a0. PMID 6095115.
- Chadd HE, Chamow SM (April 2001). "Therapeutic antibody expression technology". Current Opinion in Biotechnology. 12 (2): 188–94. doi:10.1016/S0958-1669(00)00198-1. PMID 11287236.
- Lonberg N, Huszar D (1995). "Human antibodies from transgenic mice". International Reviews of Immunology. 13 (1): 65–93. doi:10.3109/08830189509061738. PMID 7494109.
- "Bristol-Myers Buys Medarex Drugmaker for $2.4 Billion (Update3)".
- "Amgen Completes Acquisition of Abgenix; Acquisition Provides Amgen with Full Ownership of Panitumumab and Eliminates a Denosumab Royalty".
- "Archived copy". Archived from the original on June 29, 2009. Retrieved July 28, 2009.CS1 maint: archived copy as title (link)
- "Proprietary antibody platform". Archived from the original on 2013-02-03. Retrieved 2013-01-17.
- "Naturally optimized human antibodies".
- "Proprietary antibody platform".
- "Proprietary antibody platform"./
- McCafferty J, Griffiths AD, Winter G, Chiswell DJ (December 1990). "Phage antibodies: filamentous phage displaying antibody variable domains". Nature. 348 (6301): 552–4. Bibcode:1990Natur.348..552M. doi:10.1038/348552a0. PMID 2247164.
- Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G (December 1991). "By-passing immunization. Human antibodies from V-gene libraries displayed on phage". Journal of Molecular Biology. 222 (3): 581–97. doi:10.1016/0022-2836(91)90498-U. PMID 1748994.
- Carmen S, Jermutus L (July 2002). "Concepts in antibody phage display". Briefings in Functional Genomics & Proteomics. 1 (2): 189–203. doi:10.1093/bfgp/1.2.189. PMID 15239904.
- Osbourn JK (2002). "Proximity-guided (ProxiMol) antibody selection". Antibody Phage Display. Methods Mol. Biol. 178. pp. 201–5. doi:10.1385/1-59259-240-6:201. ISBN 978-1-59259-240-1. PMID 11968489.
- Abeloff MD, Armitage JO, Niederhuber JE, Kastan MB, McKenna G (2008). "Therapeutic Antibodies and Immunologic Conjugates". Abeloff's Clinical Oncology (4th ed.). Elsevier.
- "Cambridge Antibody Technology". Archived from the original on 2011-09-28.
- Osbourn JK, Derbyshire EJ, Vaughan TJ, Field AW, Johnson KS (January 1998). "Pathfinder selection: in situ isolation of novel antibodies". Immunotechnology. 3 (4): 293–302. doi:10.1016/S1380-2933(97)10007-0. PMID 9530562.
- "The Current State of Proteomic Technology". Archived from the original on 2011-10-08. Retrieved 2009-07-28.
- PhRMA Reports Identifies More than 400 Biotech Drugs in Development. Pharmaceutical Technology, August 24, 2006. Retrieved 2006-09-04.
- Hernandez I, Bott SW, Patel AS, Wolf CG, Hospodar AR, Sampathkumar S, Shrank WH (February 2018). "Pricing of monoclonal antibody therapies: higher if used for cancer?". The American Journal of Managed Care. 24 (2): 109–112. PMID 29461857.
- Breedveld FC (February 2000). "Therapeutic monoclonal antibodies". Lancet. 355 (9205): 735–40. doi:10.1016/S0140-6736(00)01034-5. PMID 10703815.
- Australian Prescriber (2006). "Monoclonal antibody therapy for non-malignant disease". Australian Prescriber. 29 (5): 130–133. doi:10.18773/austprescr.2006.079.
- Modified from Carter P (November 2001). "Improving the efficacy of antibody-based cancer therapies". Nature Reviews. Cancer. 1 (2): 118–29. doi:10.1038/35101072. PMID 11905803.
- Takimoto CH, Calvo E. (January 01, 2005) "Principles of Oncologic Pharmacotherapy" in Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (Eds) Cancer Management
- Rang HP (2003). Pharmacology. Edinburgh: Churchill Livingstone. pp. 241, for the examples infliximab, basiliximab, abciximab, daclizumab, palivusamab, gemtuzumab, alemtuzumab and rituximab, and mechanism and mode. ISBN 978-0-443-07145-4.
- Staff, Adis Insight. Bavituximab profile Last updated Jan 27 2016
- "Monoclonal antibodies to treat cancer | American Cancer Society". www.cancer.org. Retrieved 2018-04-19.
- "Monoclonal antibody drugs for cancer: How they work". Mayo Clinic. Retrieved 2018-04-19.
- "Monoclonal Antibodies: List, Types, Side Effects & FDA Uses (Cancer)". MedicineNet. Retrieved 2018-04-19.
Further reading
- 2019 Historical overview of monoclonal antibodies in the journal Nature
- Monoclonal Antibodies, from John W. Kimball's online biology textbook
External links
- Monoclonal+antibodies at the US National Library of Medicine Medical Subject Headings (MeSH)
- Antibodypedia, open-access virtual repository publishing data and commentary on any antibodies available to the scientific community.
- Antibody Purification Handbook
| Library resources about Monoclonal antibody |
Engineered monoclonal antibodies and antibody mimetics | ||
|---|---|---|
| Whole antibody | 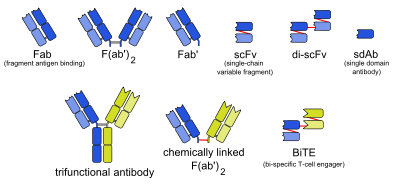 | |
| Fab fragment | ||
| Variable fragment | ||
| Smaller units | ||
| Intracellular | ||
| Antibody mimetics | ||