Ichthyosis prematurity syndrome
Ichthyosis prematurity syndrome (IPS) is a dermatological disease with known genetic causes. This syndrome is a rare subcategory of autosomal recessive congenital ichthyosis (ARCI).[1] It is associated with complications in the mid-trimester of a pregnancy leading to premature births.[2] Although most prevalent in individuals of Scandinavian origin, there have also been scattered cases in people of Japanese, Italian and Indian ethnicity.[1][3][4] This disorder is also referred to as ichthyosis congenital type IV.[1]
| Ichthyosis prematurity syndrome | |
|---|---|
| Specialty | Dermatology |
Signs and symptoms
The symptoms associated with the disorder are often confused for other dermatological disorders. The symptoms below are ones specifically associated with IPS.
Premature birth
Pregnancies that have a foetus affected with this syndrome are complicated because of polyhydramnion. Complications arise because of opaque amnionic fluid resulting from the shedding of skin. As a result, ultrasounds are difficult to conduct.[1] Triggered by the harsh environment in the uterus, delivery results around 30– 34 weeks of gestation (pregnancy) and the baby is born in prematurely.[1]
Thick caseous layer of skin
A white layer of thick scaly substance covers the surface of the skin of the infant. This is characterized by aggregating membranes in the upper layer of skin on epidermal cells.[1][5]
Desquamating skin
Red endemic skin as well as spongy and desquamating (peeling) skin are consistent with this syndrome.[1]
Respiratory problems
After premature delivery, the baby often has neonatal asphyxia from breathing amnionic debris mainly composed of shedding skin cells.[6] Neonatal refers to the very young infant and amniotic fluid refers to the liquid environment the baby is bathed in inside the uterus.
Eosinophilia
Eosinophils are a kind of a white blood cell which help protect the body from certain infections and involved in allergic responses. Eosionphelia is an abnormal increase of eosinophils in tissue, blood or both and is present in individuals born with this syndrome.[7]
Genetics
Mode of inheritance
This rare syndrome is associated with an autosomal recessive mode of inheritance. The parents are classified as heterozygote carriers of the disease and the chance that the offspring will be affected is 25%. Both parents need to have the mutant allele in order to pass on the disease to their offspring and the offspring need to inherit both mutant alleles in order to express the disorder.
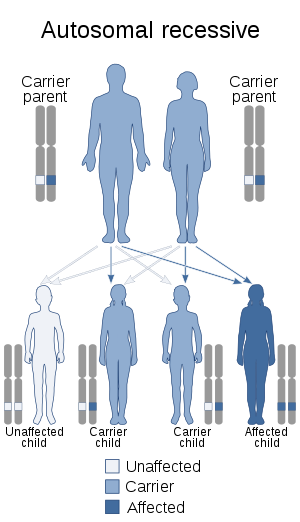
Genetic cause
IPS is caused by a number of mutations at different loci of the FATP4 gene. One nonsense mutation and a number of missense mutation in the FATP4 (SLC27A4) gene which codes for the fatty acid transport protein 4 are associated with IPS.[1][2] Splice site mutations have also been linked to IPS. When the splice site between exons and introns is mutated, it leads to a deletion or duplication event which would change the stability or soundness of the protein the RNA is coding for.
When a mutation is present in the gene coding for this protein, the protein product is not stable or able to perform its role. As a result, the pathway it is involved in is compromised and a diseased phenotype is often noted. In this particular case the fatty acid transport protein 4 is deformed due to the presence of mutations in the FATP4 gene and an interference occurs in the lipid metabolic pathway in the skin that this protein is involved with.[5]
This results in the abnormal formation of epidermal skin cells and a compromised formation and maintenance of an epidermal barrier.[5] These underlying genetic causes explain the symptoms of the flaky, dry, cutaneous skin phenotype expressed.[1]
IPS genes segregate independently of the already known general ARCI locus. As a result, IPS has been identified to have its own gene locus making it easier to diagnose.[1]
Diagnosis
Diagnosis can be made at birth by identifying the symptoms of the child. Ultrastructural diagnosis where tissues are analyzed is using electron microscopy is also conducted. A specimen of skin is obtained via a skin biopsy and analyzed to see any tell tale characteristics.[5]Genetic testing can also be done to identify the mutation on the FATP4 gene associated with fatty acid synthesis.[5] Genetic consultation through a genetic counsellor is done to determine whether the individual has this syndrome and reduces the chances of misdiagnoses with other cutaneous diseases.[2]
Treatment
The infant is intubated post delivery to stabilize the respiratory problems experienced. Often the skin condition becomes less severe resolving itself to flaky dry skin as the individual grows. No intervention is usually required and the condition becomes less severe as the patient grows. The dry skin symptoms can be managed with topical ointments or creams and the individual remains otherwise healthy.[6]
Prognosis
There are no life-threatening complications after the perinatal period (around the time of birth) and the skin conditions persist but to a lesser degree of severity. Individuals have a favourable prognosis as symptoms can be managed and past the infancy stage are not life-threatening. The red skin edema improves after a three-week period but the ichthyosis scaling persists.[6] Asthma has been recorded in some cases later on in the individual's life and sign of atopic dermatitis persist, follicular hyperkeratosis and small amounts of scaling at the scalp that goes on into adulthood but otherwise the individual continues a healthy life.[6]
Epidemiology
Frequencies of this disease are the greatest in Norway with a few Finnish cases have also having been noted to date.[6] Some cases have been found in other ethnicities such as in people of Indian or Japanese descent as well as a north Italian family. These cases are scattered and there are potentially more under reported cases as this disease is often under diagnosed for other cutaneous diseases. It is most prevalent in a defined region in the middle of Norway and Sweden with a heterozygote carrier frequency of 1 in 50.[6]
References
- Klar, J; Gedde-Dahl, T; Larsson, M; et al. (2004). "Assignment of the locus for ichthyosis prematurity syndrome to chromosome 9q33.3-34.13". J Med Genet. 41 (3): 208–212. doi:10.1136/jmg.2003.012567. PMC 1735696. PMID 14985385.
- "Ichthyosis Prematurity Syndrome". OMIM. Retrieved 3 December 2015.
- Ikuya, Tsuge; Masashi, Morishita; Takema, Kato; et al. (2015). "Identification of novel FATP4 mutations in a Japanese patient with ichthyosis prematurity syndrome". Human Genome Variation. 2: 15003. doi:10.1038/hgv.2015.3. PMC 4785586. PMID 27081519.
- Renu, George (2014). "Ichthyosis prematurity syndrome: A case report from India". Journal of the American Academy of Dermatology. 70 (5): AB145. doi:10.1016/j.jaad.2014.01.603. Retrieved 16 October 2015.
- Sobol, Maria; Dahl, Niklas; Klar, Joakim (2011). "FATP4 missense and nonsense mutations cause similar features in Ichthyosis Prematurity Syndrome". BMC Research Notes. 4: 90. doi:10.1186/1756-0500-4-90. PMC 3072334. PMID 21450060.
- Bygum, Anette; Westermark, Per; Brandrup, Flemming (2008). "Ichthyosis prematurity syndrome: A well-defined congenital ichthyosis subtype". Journal of the American Academy of Dermatology. 59 (5): S71–S74. doi:10.1016/j.jaad.2008.06.014. PMID 19119129.
- "Eosinophelia". Mount Sinai Hospital. Retrieved 9 November 2015.
External links
| Classification | |
|---|---|
| External resources |