Glucose-6-phosphate dehydrogenase
Glucose-6-phosphate dehydrogenase (G6PD or G6PDH) (EC 1.1.1.49) is a cytosolic enzyme that catalyzes the chemical reaction
- D-glucose 6-phosphate + NADP+ + H2O ⇌ 6-phospho-D-glucono-1,5-lactone + NADPH + H+
| Glucose-6-phosphate dehydrogenase, NAD binding domain | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 glucose 6-phosphate dehydrogenase from leuconostoc mesenteroides | |||||||||
| Identifiers | |||||||||
| Symbol | G6PD_N | ||||||||
| Pfam | PF00479 | ||||||||
| Pfam clan | CL0063 | ||||||||
| InterPro | IPR022674 | ||||||||
| PROSITE | PDOC00067 | ||||||||
| SCOPe | 1dpg / SUPFAM | ||||||||
| |||||||||
| Glucose-6-phosphate dehydrogenase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC number | 1.1.1.49 | ||||||||
| CAS number | 9001-40-5 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
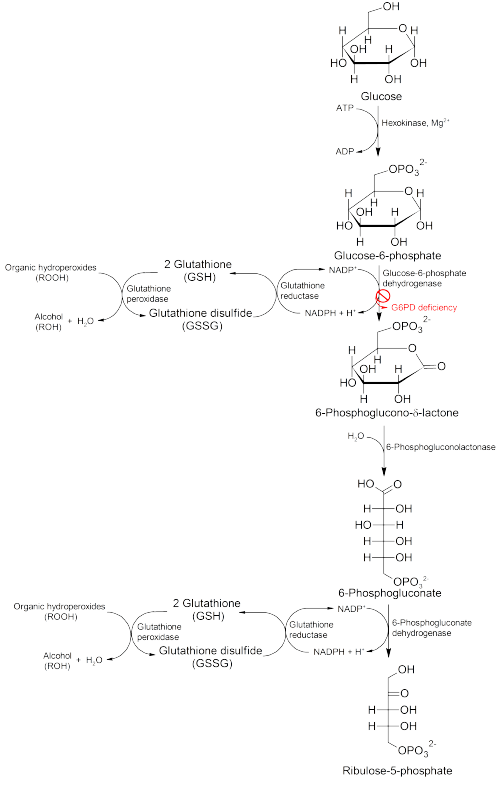
This enzyme participates in the pentose phosphate pathway (see image), a metabolic pathway that supplies reducing energy to cells (such as erythrocytes) by maintaining the level of the co-enzyme nicotinamide adenine dinucleotide phosphate (NADPH). The NADPH in turn maintains the level of glutathione in these cells that helps protect the red blood cells against oxidative damage from compounds like hydrogen peroxide.[1] Of greater quantitative importance is the production of NADPH for tissues involved in biosynthesis of fatty acids or isoprenoids, such as the liver, mammary glands, adipose tissue, and the adrenal glands. G6PD reduces NADP+ to NADPH while oxidizing glucose-6-phosphate.[2]
Clinically, an X-linked genetic deficiency of G6PD predisposes a person to non-immune hemolytic anemia.[3]
Species distribution
G6PD is widely distributed in many species from bacteria to humans. Multiple sequence alignment of over 100 known G6PDs from different organisms reveal sequence identity ranging from 30% to 94%.[4] Human G6PD has over 30% identity in amino acid sequence to G6PD sequences from other species.[5] Humans also have two isoforms of a single gene coding for G6PD.[6] Moreover, 150 different human G6PD mutants have been documented.[4] These mutations are mainly missense mutations that result in amino acid substitutions,[7] and while some of them result in G6PD deficiency, others do not seem to result in any noticeable functional differences.[7] Some scientists have proposed that some of the genetic variation in human G6PD resulted from generations of adaptation to malarial infection.[8]
Other species experience a variation in G6PD as well. In higher plants, several isoforms of G6PDH have been reported, which are localized in the cytosol, the plastidic stroma, and peroxisomes.[9] A modified F420-dependent (as opposed to NADP+-dependent) G6PD is found in Mycobacterium tuberculosis, and is of interest for treating tuberculosis.[10] The bacterial G6PD found in Leuconostoc mesenteroides was shown to be reactive toward 4-Hydroxynonenal, in addition to G6P.[11]
Enzyme structure
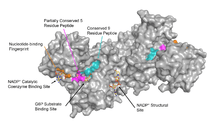
G6PD is generally found as a dimer of two identical monomers (see main thumbnail).[7] Depending on conditions, such as pH, these dimers can themselves dimerize to form tetramers.[5] Each monomer in the complex has a substrate binding site that binds to G6P, and a catalytic coenzyme binding site that binds to NADP+/NADPH using the Rossman fold.[4] For some higher organisms, such as humans, G6PD contains an additional NADP+ binding site, called the NADP+ structural site, that does not seem to participate directly in the reaction catalyzed by G6PD. The evolutionary purpose of the NADP+ structural site is unknown.[4] As for size, each monomer is approximately 500 amino acids long (514 amino acids for humans[5]).
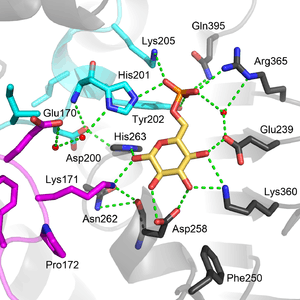
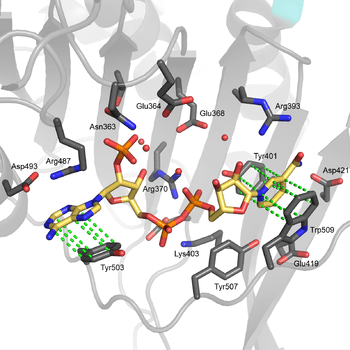
Functional and structural conservation between human G6PD and Leuconostoc mesenteroides G6PD points to 3 widely conserved regions on the enzyme: a 9 residue peptide in the substrate binding site, RIDHYLGKE (residues 198-206 on human G6PD), a nucleotide-binding fingerprint, GxxGDLA (residues 38-44 on human G6PD), and a partially conserved sequence EKPxG near the substrate binding site (residues 170-174 on human G6PD), where we have use "x" to denote a variable amino acid.[4] The crystal structure of G6PD reveals an extensive network of electrostatic interactions and hydrogen bonding involving G6P, 3 water molecules, 3 lysines, 1 arginine, 2 histidines, 2 glutamic acids, and other polar amino acids.
The proline at position 172 is thought to play a crucial role in positioning Lys171 correctly with respect to the substrate, G6P. In the two crystal structures of normal human G6P, Pro172 is seen exclusively in the cis confirmation, while in the crystal structure of one disease causing mutant (variant Canton R459L), Pro172 is seen almost exclusively in the trans confirmation.[4]
With access to crystal structures, some scientists have tried to model the structures of other mutants. For example, in German ancestry, where enzymopathy due to G6PD deficiency is rare, mutation sites on G6PD have been shown to lie near the NADP+ binding site, the G6P binding site, and near the interface between the two monomers. Thus, mutations in these critical areas are possible without completely disrupting the function of G6PD.[7] In fact, it has been shown that most disease causing mutations of G6PD occur near the NADP+ structural site.[12]
NADP+ structural site
The NADP+ structural site is located greater than 20Å away from the substrate binding site and the catalytic coenzyme NADP+ binding site. Its purpose in the enzyme catalyzed reaction has been unclear for many years. For some time, it was thought that NADP+ binding to the structural site was necessary for dimerization of the enzyme monomers. However, this was shown to be incorrect.[12] On the other hand, it was shown that the presence of NADP+ at the structural site promotes the dimerization of dimers to form enzyme tetramers.[12] It was also thought that the tetramer state was necessary for catalytic activity; however, this too was shown to be false.[12] The NADP+ structural site is quite different from the NADP+ catalytic coenzyme binding site, and contains the nucleotide-binding fingerprint.
The structural site bound to NADP+ possesses favorable interactions that keep it tightly bound. In particular, there is a strong network of hydrogen bonding with electrostatic charges being diffused across multiple atoms through hydrogen bonding with 4 water molecules (see figure). Moreover, there is an extremely strong set of hydrophobic stacking interactions that result in overlapping π systems.
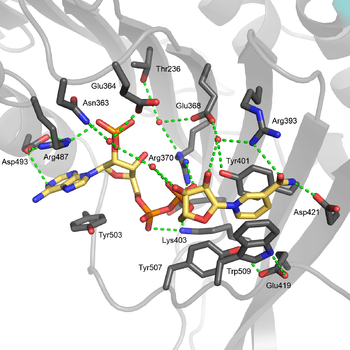
The structural site has been shown to be important for maintaining the long term stability of the enzyme.[12] More than 40 severe class I mutations involve mutations near the structural site, thus affecting the long term stability of these enzymes in the body, ultimately resulting in G6PD deficiency.[12] For example, two severe class I mutations, G488S and G488V, drastically increase the dissociation constant between NADP+ and the structural site by a factor of 7 to 13. With the proximity of residue 488 to Arg487, it is thought that a mutation at position 488 could affect the positioning of Arg487 relative to NADP+,[12] and thus disrupt binding.
Regulation
G6PD converts G6P into 6-phosphoglucono-δ-lactone and is the rate-limiting enzyme of the pentose phosphate pathway. Thus, regulation of G6PD has downstream consequences for the activity of the rest of the pentose phosphate pathway.
Glucose-6-phosphate dehydrogenase is stimulated by its substrate G6P. The usual ratio of NADPH/NADP+ in the cytosol of tissues engaged in biosyntheses is about 100/1. Increased utilization of NADPH for fatty acid biosynthesis will dramatically increase the level of NADP+, thus stimulating G6PD to produce more NADPH. Yeast G6PD is inhibited by long chain fatty acids according to two older publications[13][14] and might be product inhibition in fatty acid synthesis which requires NADPH.
G6PD is negatively regulated by acetylation on lysine 403 (Lys403), an evolutionarily conserved residue. The K403 acetylated G6PD is incapable of forming active dimers and displays a complete loss of activity. Mechanistically, acetylating Lys304 sterically hinders the NADP+ from entering the NADP+ structural site, which reduces the stability of the enzyme. Cells sense extracellular oxidative stimuli to decrease G6PD acetylation in a SIRT2-dependent manner. The SIRT2-mediated deacetylation and activation of G6PD stimulates pentose phosphate pathway to supply cytosolic NADPH to counteract oxidative damage and protect mouse erythrocytes.[15]
Regulation can also occur through genetic pathways. The isoform, G6PDH, is regulated by transcription and posttranscription factors.[16] Moreover, G6PD is one of a number of glycolytic enzymes activated by the transcription factor hypoxia-inducible factor 1 (HIF1).[17]
Clinical significance
G6PD is remarkable for its genetic diversity. Many variants of G6PD, mostly produced from missense mutations, have been described with wide-ranging levels of enzyme activity and associated clinical symptoms. Two transcript variants encoding different isoforms have been found for this gene.[18]
Glucose-6-phosphate dehydrogenase deficiency is very common worldwide, and causes acute hemolytic anemia in the presence of simple infection, ingestion of fava beans, or reaction with certain medicines, antibiotics, antipyretics, and antimalarials.[3]
Cell growth and proliferation are affected by G6PD.[19] G6PD inhibitors are under investigation to treat cancers and other conditions.[17] In vitro cell proliferation assay indicates that G6PD inhibitors, DHEA (dehydroepiandrosterone) and ANAD (6-aminonicotinamide), effectively decrease the growth of AML cell lines.[19][20] G6PD is hypomethylated at K403 in acute myeloid leukemia, SIRT2 activates G6PD to enhance NADPH production and promote leukemia cell proliferation.[20]
See also
- Glucose-6-phosphate dehydrogenase deficiency
- Genetic resistance to malaria
References
- Thomas D, Cherest H, Surdin-Kerjan Y (March 1991). "Identification of the structural gene for glucose-6-phosphate dehydrogenase in yeast. Inactivation leads to a nutritional requirement for organic sulfur". The EMBO Journal. 10 (3): 547–53. PMC 452682. PMID 2001672.
- Aster J, Kumar V, Robbins SL, Abbas AK, Fausto N, Cotran RS (2010). Robbins and Cotran Pathologic Basis of Disease. Saunders/Elsevier. pp. Kindle Locations 33340–33341. ISBN 1-4160-3121-9.
- Cappellini MD, Fiorelli G (January 2008). "Glucose-6-phosphate dehydrogenase deficiency". Lancet. 371 (9606): 64–74. doi:10.1016/S0140-6736(08)60073-2. PMID 18177777.
- Kotaka M, Gover S, Vandeputte-Rutten L, Au SW, Lam VM, Adams MJ (May 2005). "Structural studies of glucose-6-phosphate and NADP+ binding to human glucose-6-phosphate dehydrogenase" (PDF). Acta Crystallographica D. 61 (Pt 5): 495–504. doi:10.1107/S0907444905002350. PMID 15858258.
- Au SW, Gover S, Lam VM, Adams MJ (March 2000). "Human glucose-6-phosphate dehydrogenase: the crystal structure reveals a structural NADP(+) molecule and provides insights into enzyme deficiency". Structure. 8 (3): 293–303. doi:10.1016/S0969-2126(00)00104-0. PMID 10745013.
- "G6PD glucose-6-phosphate dehydrogenase [ Homo sapiens (human) ]". NCBI. Retrieved 13 December 2015.
- Kiani F, Schwarzl S, Fischer S, Efferth T (July 2007). "Three-dimensional modeling of glucose-6-phosphate dehydrogenase-deficient variants from German ancestry". PLOS One. 2 (7): e625. doi:10.1371/journal.pone.0000625. PMC 1913203. PMID 17637841.
- Luzzatto L, Bienzle U (June 1979). "The malaria/G.-6-P.D. hypothesis". Lancet. 1 (8127): 1183–4. doi:10.1016/S0140-6736(79)91857-9. PMID 86896.
- Corpas FJ, Barroso JB, Sandalio LM, Distefano S, Palma JM, Lupiáñez JA, Del Río LA (March 1998). "A dehydrogenase-mediated recycling system of NADPH in plant peroxisomes". The Biochemical Journal. 330 (Pt 2): 777–84. doi:10.1042/bj3300777. PMC 1219205. PMID 9480890.
- Bashiri G, Squire CJ, Moreland NJ, Baker EN (June 2008). "Crystal structures of F420-dependent glucose-6-phosphate dehydrogenase FGD1 involved in the activation of the anti-tuberculosis drug candidate PA-824 reveal the basis of coenzyme and substrate binding". The Journal of Biological Chemistry. 283 (25): 17531–41. doi:10.1074/jbc.M801854200. PMID 18434308.
- Szweda LI, Uchida K, Tsai L, Stadtman ER (February 1993). "Inactivation of glucose-6-phosphate dehydrogenase by 4-hydroxy-2-nonenal. Selective modification of an active-site lysine". The Journal of Biological Chemistry. 268 (5): 3342–7. PMID 8429010.
- Wang XT, Chan TF, Lam VM, Engel PC (August 2008). "What is the role of the second "structural" NADP+-binding site in human glucose 6-phosphate dehydrogenase?". Protein Science. 17 (8): 1403–11. doi:10.1110/ps.035352.108. PMC 2492815. PMID 18493020.
- Eger-Neufeldt I, Teinzer A, Weiss L, Wieland O (March 1965). "Inhibition of glucose-6-phosphate dehydrogenase by long chain acyl-coenzyme A". Biochemical and Biophysical Research Communications. 19 (1): 43–48. doi:10.1016/0006-291X(65)90116-6.
- Kawaguchi A, Bloch K (September 1974). "Inhibition of glucose 6-phosphate dehydrogenase by palmitoyl coenzyme A". The Journal of Biological Chemistry. 249 (18): 5793–800. PMID 4153382.
- Wang YP, Zhou LS, Zhao YZ, Wang SW, Chen LL, Liu LX, Ling ZQ, Hu FJ, Sun YP, Zhang JY, Yang C, Yang Y, Xiong Y, Guan KL, Ye D (June 2014). "Regulation of G6PD acetylation by SIRT2 and KAT9 modulates NADPH homeostasis and cell survival during oxidative stress". The EMBO Journal. 33 (12): 1304–20. doi:10.1002/embj.201387224. PMC 4194121. PMID 24769394.
- Kletzien RF, Harris PK, Foellmi LA (February 1994). "Glucose-6-phosphate dehydrogenase: a "housekeeping" enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress". FASEB Journal. 8 (2): 174–81. PMID 8119488.
- de Lartigue J (2012-06-12). "Cancer Research Moves Beyond the Original Hallmarks of Cancer". OncLive.
- "Entrez Gene: G6PD glucose-6-phosphate dehydrogenase".
- Tian WN, Braunstein LD, Pang J, Stuhlmeier KM, Xi QC, Tian X, Stanton RC (April 1998). "Importance of glucose-6-phosphate dehydrogenase activity for cell growth". The Journal of Biological Chemistry. 273 (17): 10609–17. doi:10.1074/jbc.273.17.10609. PMID 9553122.
- Xu SN, Wang TS, Li X, Wang YP (September 2016). "SIRT2 activates G6PD to enhance NADPH production and promote leukaemia cell proliferation". Scientific Reports. 6: 32734. doi:10.1038/srep32734. PMC 5009355. PMID 27586085.
Further reading
- Vulliamy T, Beutler E, Luzzatto L (1993). "Variants of glucose-6-phosphate dehydrogenase are due to missense mutations spread throughout the coding region of the gene". Human Mutation. 2 (3): 159–67. doi:10.1002/humu.1380020302. PMID 8364584.
- Mason PJ (September 1996). "New insights into G6PD deficiency". British Journal of Haematology. 94 (4): 585–91. PMID 8826878.
- Wajcman H, Galactéros F (August 2004). "[Glucose 6-phosphate dehydrogenase deficiency: a protection against malaria and a risk for hemolytic accidents]". Comptes Rendus Biologies (in French). 327 (8): 711–20. doi:10.1016/j.crvi.2004.07.010. PMID 15506519.
External links
PDB gallery | |
|---|---|
|