Genetic engineering techniques
Genetic engineering can be accomplished using multiple techniques. There are a number of steps that are followed before a genetically modified organism (GMO) is created. Genetic engineers must first choose what gene they wish to insert, modify, or delete. The gene must then be isolated and incorporated, along with other genetic elements, into a suitable vector. This vector is then used to insert the gene into the host genome, creating a transgenic or edited organism. The ability to genetically engineer organisms is built on years of research and discovery on how genes function and how we can manipulate them. Important advances included the discovery of restriction enzymes and DNA ligases and the development of polymerase chain reaction and sequencing.
| Part of a series on |
| Genetic engineering |
|---|
| Genetically modified organisms |
| History and regulation |
| Process |
| Applications |
| Controversies |
This allowed the gene of interest to be isolated and then incorporated into a vector. Often a promoter and terminator region was added as well as a selectable marker gene. The gene may be modified further at this point to make it express more efficiently. This vector is then inserted into the host organism's genome. For animals, the gene is typically inserted into embryonic stem cells, while in plants it can be inserted into any tissue that can be cultured into a fully developed plant. Common techniques include microinjection, virus-mediated, Agrobacterium-mediated or biolistics. Further tests are carried out on the resulting organism to ensure stable integration, inheritance and expression. First generation offspring are heterozygous, requiring them to be inbred to create the homozygous pattern necessary for stable inheritance. Homozygosity must be confirmed in second generation specimens.
Traditional techniques inserted the genes randomly into the host's genome. Advances have allowed genes to be inserted at specific locations within a genome, which reduces the unintended side effects of random insertion. Early targeting systems relied on meganucleases and zinc finger nucleases. Since 2009 more accurate and easier systems to implement have been developed. Transcription activator-like effector nucleases (TALENs) and the Cas9-guideRNA system (adapted from CRISPR) are the two most commonly used. They may potentially be useful in gene therapy and other procedures that require accurate or high throughput targeting.
History
Many different discoveries and advancements led to the development of genetic engineering. Human-directed genetic manipulation began with the domestication of plants and animals through artificial selection in about 12,000 BC.[1]:1 Various techniques were developed to aid in breeding and selection. Hybridization was one way rapid changes in an organism's genetic makeup could be introduced. Crop hybridization most likely first occurred when humans began growing genetically distinct individuals of related species in close proximity.[2]:32 Some plants were able to be propagated by vegetative cloning.[2]:31
Genetic inheritance was first discovered by Gregor Mendel in 1865, following experiments crossing peas.[3] In 1928 Frederick Griffith proved the existence of a "transforming principle" involved in inheritance, which was identified as DNA in 1944 by Oswald Avery, Colin MacLeod, and Maclyn McCarty. Frederick Sanger developed a method for sequencing DNA in 1977, greatly increasing the genetic information available to researchers.
After discovering the existence and properties of DNA, tools had to be developed that allowed it to be manipulated. In 1970 Hamilton Smiths lab discovered restriction enzymes, enabling scientists to isolate genes from an organism's genome.[4] DNA ligases, which join broken DNA together, were discovered earlier in 1967.[5] By combining the two enzymes it became possible to "cut and paste" DNA sequences to create recombinant DNA. Plasmids, discovered in 1952,[6] became important tools for transferring information between cells and replicating DNA sequences. Polymerase chain reaction (PCR), developed by Kary Mullis in 1983, allowed small sections of DNA to be amplified (replicated) and aided identification and isolation of genetic material.
As well as manipulating DNA, techniques had to be developed for its insertion into an organism's genome. Griffith's experiment had already shown that some bacteria had the ability to naturally uptake and express foreign DNA. Artificial competence was induced in Escherichia coli in 1970 by treating them with calcium chloride solution (CaCl2).[7] Transformation using electroporation was developed in the late 1980s, increasing the efficiency and bacterial range.[8] In 1907 a bacterium that caused plant tumors, Agrobacterium tumefaciens, had been discovered. In the early 1970s it was found that this bacteria inserted its DNA into plants using a Ti plasmid.[9] By removing the genes in the plasmid that caused the tumor and adding in novel genes, researchers were able to infect plants with A. tumefaciens and let the bacteria insert their chosen DNA into the genomes of the plants.[10]
Choosing target genes
The first step is to identify the target gene or genes to insert into the host organism. This is driven by the goal for the resultant organism. In some cases only one or two genes are affected. For more complex objectives entire biosynthetic pathways involving multiple genes may be involved. Once found genes and other genetic information from a wide range of organisms can be inserted into bacteria for storage and modification, creating genetically modified bacteria in the process. Bacteria are cheap, easy to grow, clonal, multiply quickly, relatively easy to transform and can be stored at -80 °C almost indefinitely. Once a gene is isolated it can be stored inside the bacteria providing an unlimited supply for research.[11]
Genetic screens can be carried out to determine potential genes followed by other tests identify the best candidates. A simple screen involves randomly mutating DNA with chemicals or radiation and then selecting those that display the desired trait. For organisms where mutation is not practical, scientists instead look for individuals among the population who present the characteristic through naturally-occurring mutations. Processes that look at a phenotype and then try and identify the gene responsible are called forward genetics. The gene then needs to be mapped by comparing the inheritance of the phenotype with known genetic markers. Genes that are close together are likely to be inherited together.[12]
Another option is reverse genetics. This approach involves targeting a specific gene with a mutation and then observing what phenotype develops.[12] The mutation can be designed to inactivate the gene or only allow it to become active only under certain conditions. Conditional mutations are useful for identifying genes that are normally lethal if non-functional.[13] As genes with similar functions share similar sequences (homologous) it is possible to predict the likely function of a gene by comparing its sequence to that of well-studied genes from model organisms.[12] The development of microarrays, transcriptomes and genome sequencing has made it much easier to find desirable genes.[14]
The bacteria Bacillus thuringiensis was first discovered in 1901 as the causative agent in the death of silkworms. Due to these insecticidal properties, the bacteria was used as a biological insecticide, developed commercially in 1938. The cry proteins were discovered to provide the insecticidal activity in 1956, and by the 1980s, scientists had successfully cloned the gene that encodes this protein and expressed it in plants.[15] The gene that provides resistance to the herbicide glyphosate was found after seven years of searching in bacteria that living in the outflow pipe of a Monsanto RoundUp manufacturing facility.[16] In animals, the majority of genes used are growth hormone genes.[17]
Gene manipulation
All genetic engineering processes involve the modification of DNA. Traditionally DNA was isolated from the cells of organisms. Later, genes came to be cloned from a DNA segment after the creation of a DNA library or artificially synthesised. Once isolated, additional genetic elements are added to the gene to allow it to be expressed in the host organism and to aid selection.
Extraction from cells
First the cell must be gently opened, exposing the DNA without causing too much damage to it. The methods used vary depending on the type of cell. Once open, the DNA must be separated from the other cellular components. A ruptured cell contains proteins and other cell debris. By mixing with phenol and/or chloroform, followed by centrifuging, the nucleic acids can be separated from this debris into an upper aqueous phase. This aqueous phase can be removed and further purified if necessary by repeating the phenol-chloroform steps. The nucleic acids can then be precipitated from the aqueous solution using ethanol or isopropanol. Any RNA can be removed by adding a ribonuclease that will degrade it. Many companies now sell kits that simplify the process.[18]
Gene isolation
The gene of interest must be separated from the extracted DNA. If the sequence is not known then a common method is to break the DNA up with a random digestion method. This is usually accomplished using restriction enzymes (enzymes that cut DNA). A partial restriction digest cuts only some of the restriction sites, resulting in overlapping DNA fragment segments. The DNA fragments are put into individual plasmid vectors and grown inside bacteria. Once in the bacteria the plasmid is copied as the bacteria divides. To determine if a useful gene is present on a particular fragment the DNA library is screened for the desired phenotype. If the phenotype is detected then it is possible that the bacteria contains the target gene.
If the gene does not have a detectable phenotype or a DNA library does not contain the correct gene, other methods must be used to isolate it. If the position of the gene can be determined using molecular markers then chromosome walking is one way to isolate the correct DNA fragment. If the gene expresses close homology to a known gene in another species, then it could be isolated by searching for genes in the library that closely match the known gene.[19]
For known DNA sequences, restriction enzymes that cut the DNA on either side of the gene can be used. Gel electrophoresis then sorts the fragments according to length.[20] Some gels can separate sequences that differ by a single base-pair. The DNA can be visualised by staining it with ethidium bromide and photographing under UV light. A marker with fragments of known lengths can be laid alongside the DNA to estimate the size of each band. The DNA band at the correct size should contain the gene, where it can be excised from the gel.[18]:40–41 Another technique to isolate genes of known sequences involves polymerase chain reaction (PCR).[21] PCR is a powerful tool that can amplify a given sequence, which can then be isolated through gel electrophoresis. Its effectiveness drops with larger genes and it has the potential to introduce errors into the sequence.
It is possible to artificially synthesise genes.[22] Some synthetic sequences are available commercially, forgoing many of these early steps.[23]
Modification
The gene to be inserted must be combined with other genetic elements in order for it to work properly. The gene can be modified at this stage for better expression or effectiveness. As well as the gene to be inserted most constructs contain a promoter and terminator region as well as a selectable marker gene. The promoter region initiates transcription of the gene and can be used to control the location and level of gene expression, while the terminator region ends transcription. A selectable marker, which in most cases confers antibiotic resistance to the organism it is expressed in, is used to determine which cells are transformed with the new gene. The constructs are made using recombinant DNA techniques, such as restriction digests, ligations and molecular cloning.[24]
Inserting DNA into the host genome
Once the gene is constructed it must be stably integrated into the target organisms genome or exist as extrachromosomal DNA. There are a number of techniques available for inserting the gene into the host genome and they vary depending on the type of organism targeted. In multicellular eukaryotes, if the transgene is incorporated into the host's germline cells, the resulting host cell can pass the transgene to its progeny. If the transgene is incorporated into somatic cells, the transgene can not be inherited.[25]
Transformation
Transformation is the direct alteration of a cell's genetic components by passing the genetic material through the cell membrane. About 1% of bacteria are naturally able to take up foreign DNA, but this ability can be induced in other bacteria.[26] Stressing the bacteria with a heat shock or electroporation can make the cell membrane permeable to DNA that may then be incorporated into the genome or exist as extrachromosomal DNA. Typically the cells are incubated in a solution containing divalent cations (often calcium chloride) under cold conditions, before being exposed to a heat pulse (heat shock). Calcium chloride partially disrupts the cell membrane, which allows the recombinant DNA to enter the host cell. It is suggested that exposing the cells to divalent cations in cold condition may change or weaken the cell surface structure, making it more permeable to DNA. The heat-pulse is thought to create a thermal imbalance across the cell membrane, which forces the DNA to enter the cells through either cell pores or the damaged cell wall. Electroporation is another method of promoting competence. In this method the cells are briefly shocked with an electric field of 10-20 kV/cm, which is thought to create holes in the cell membrane through which the plasmid DNA may enter. After the electric shock, the holes are rapidly closed by the cell's membrane-repair mechanisms. Up-taken DNA can either integrate with the bacterials genome or, more commonly, exist as extrachromosomal DNA.

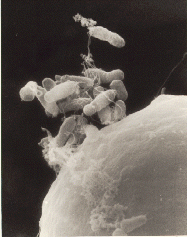
In plants the DNA is often inserted using Agrobacterium-mediated recombination,[27] taking advantage of the Agrobacteriums T-DNA sequence that allows natural insertion of genetic material into plant cells.[28] Plant tissue are cut into small pieces and soaked in a fluid containing suspended Agrobacterium. The bacteria will attach to many of the plant cells exposed by the cuts. The bacteria uses conjugation to transfer a DNA segment called T-DNA from its plasmid into the plant. The transferred DNA is piloted to the plant cell nucleus and integrated into the host plants genomic DNA.The plasmid T-DNA is integrated semi-randomly into the genome of the host cell.[29]
By modifying the plasmid to express the gene of interest, researchers can insert their chosen gene stably into the plants genome. The only essential parts of the T-DNA are its two small (25 base pair) border repeats, at least one of which is needed for plant transformation.[30][31] The genes to be introduced into the plant are cloned into a plant transformation vector that contains the T-DNA region of the plasmid. An alternative method is agroinfiltration.[32][33]
Another method used to transform plant cells is biolistics, where particles of gold or tungsten are coated with DNA and then shot into young plant cells or plant embryos.[34] Some genetic material enters the cells and transforms them. This method can be used on plants that are not susceptible to Agrobacterium infection and also allows transformation of plant plastids. Plants cells can also be transformed using electroporation, which uses an electric shock to make the cell membrane permeable to plasmid DNA. Due to the damage caused to the cells and DNA the transformation efficiency of biolistics and electroporation is lower than agrobacterial transformation.
Transfection
Transformation has a different meaning in relation to animals, indicating progression to a cancerous state, so the process used to insert foreign DNA into animal cells is usually called transfection.[35] There are many ways to directly introduce DNA into animal cells in vitro. Often these cells are stem cells that are used for gene therapy. Chemical based methods uses natural or synthetic compounds to form particles that facilitate the transfer of genes into cells.[36] These synthetic vectors have the ability to bind DNA and accommodate large genetic transfers.[37] One of the simplest methods involves using calcium phosphate to bind the DNA and then exposing it to cultured cells. The solution, along with the DNA, is encaspulated by the cells.[38] Liposomes and polymers can be used as vectors to deliver DNA into cultured animal cells. Positively charged liposomes bind with DNA, while polymers can designed that interact with DNA.[36] They form lipoplexes and polyplexes respectively, which are then up-taken by the cells. Other techniques include using electroporation and biolistics.[39] In some cases, transfected cells may stably integrate external DNA into their own genome, this process is known as stable transfection.[40]
To create transgenic animals the DNA must be inserted into viable embryos or eggs. This is usually accomplished using microinjection, where DNA is injected through the cell's nuclear envelope directly into the nucleus.[26] Superovulated fertilised eggs are collected at the single cell stage and cultured in vitro. When the pronuclei from the sperm head and egg are visible through the protoplasm the genetic material is injected into one of them. The oocyte is then implanted in the oviduct of a pseudopregnant animal.[41] Another method is Embryonic Stem Cell-Mediated Gene Transfer. The gene is transfected into embryonic stem cells and then they are inserted into mouse blastocysts that are then implanted into foster mothers. The resulting offspring are chimeric, and further mating can produce mice fully transgenic with the gene of interest.[42]
Transduction
Transduction is the process by which foreign DNA is introduced into a cell by a virus or viral vector.[43] Genetically modified viruses can be used as viral vectors to transfer target genes to another organism in gene therapy.[44] First the virulent genes are removed from the virus and the target genes are inserted instead. The sequences that allow the virus to insert the genes into the host organism must be left intact. Popular virus vectors are developed from retroviruses or adenoviruses. Other viruses used as vectors include, lentiviruses, pox viruses and herpes viruses. The type of virus used will depend on the cells targeted and whether the DNA is to be altered permanently or temporarily.
Regeneration
As often only a single cell is transformed with genetic material, the organism must be regenerated from that single cell. In plants this is accomplished through the use of tissue culture.[45][46] Each plant species has different requirements for successful regeneration. If successful, the technique produces an adult plant that contains the transgene in every cell.[47] In animals it is necessary to ensure that the inserted DNA is present in the embryonic stem cells.[27] Offspring can be screened for the gene. All offspring from the first generation are heterozygous for the inserted gene and must be inbred to produce a homozygous specimen. Bacteria consist of a single cell and reproduce clonally so regeneration is not necessary. Selectable markers are used to easily differentiate transformed from untransformed cells.
Cells that have been successfully transformed with the DNA contain the marker gene, while those not transformed will not. By growing the cells in the presence of an antibiotic or chemical that selects or marks the cells expressing that gene, it is possible to separate modified from unmodified cells. Another screening method involves a DNA probe that sticks only to the inserted gene. These markers are usually present in the transgenic organism, although a number of strategies have been developed that can remove the selectable marker from the mature transgenic plant.[48]
Confirmation
Finding that a recombinant organism contains the inserted genes is not usually sufficient to ensure that they will be appropriately expressed in the intended tissues. Further testing using PCR, Southern hybridization, and DNA sequencing is conducted to confirm that an organism contains the new gene.[49] These tests can also confirm the chromosomal location and copy number of the inserted gene. Once confirmed methods that look for and measure the gene products (RNA and protein) are also used to assess gene expression, transcription, RNA processing patterns and expression and localization of protein product(s). These include northern hybridisation, quantitative RT-PCR, Western blot, immunofluorescence, ELISA and phenotypic analysis.[50] When appropriate, the organism's offspring are studied to confirm that the transgene and associated phenotype are stably inherited.
Gene targeting
Traditional methods of genetic engineering generally insert the new genetic material randomly within the host genome. This can impair or alter other genes within the organism. Methods were developed that inserted the new genetic material into specific sites within an organism genome. Early methods that targeted genes at certain sites within a genome relied on homologous recombination.[51] By creating DNA constructs that contain a template that matches the targeted genome sequence, it is possible that the HR processes within the cell will insert the construct at the desired location. Using this method on embryonic stem cells led to the development of transgenic mice with targeted knocked out. It has also been possible to knock in genes or alter gene expression patterns.[52]
If a vital gene is knocked out it can prove lethal to the organism. In order to study the function of these genes, site specific recombinases (SSR) were used. The two most common types are the Cre-LoxP and Flp-FRT systems. Cre recombinase is an enzyme that removes DNA by homologous recombination between binding sequences known as Lox-P sites. The Flip-FRT system operates in a similar way, with the Flip recombinase recognizing FRT sequences. By crossing an organism containing the recombinase sites flanking the gene of interest with an organism that expresses the SSR under control of tissue specific promoters, it is possible to knock out or switch on genes only in certain cells. This has also been used to remove marker genes from transgenic animals. Further modifications of these systems allowed researchers to induce recombination only under certain conditions, allowing genes to be knocked out or expressed at desired times or stages of development.[52]
Genome editing uses artificially engineered nucleases that create specific double-stranded breaks at desired locations in the genome. The breaks are subject to cellular DNA repair processes that can be exploited for targeted gene knock-out, correction or insertion at high frequencies. If a donor DNA containing the appropriate sequence (homologies) is present, then new genetic material containing the transgene will be integrated at the targeted site with high efficiency by homologous recombination.[53] There are four families of engineered nucleases: meganucleases,[54][55] ZFNs,[56][57] transcription activator-like effector nucleases (TALEN),[58][59] the CRISPR/Cas (clustered regularly interspaced short palindromic repeat/CRISPRassociated protein (e.g. CRISPR/Cas9).[60][61] Among the four types, TALEN and CRISPR/Cas are the two most commonly used.[62] Recent advances have looked at combining multiple systems to exploit the best features of both (e.g. megaTAL that are a fusion of a TALE DNA binding domain and a meganuclease).[63] Recent research has also focused on developing strategies to create gene knock-out or corrections without creating double stranded breaks (base editors).[62]
Meganucleases and Zinc finger nucleases
Meganucleases were first used in 1988 in mammalian cells.[64] Meganucleases are endodeoxyribonucleases that function as restriction enzymes with long recognition sites, making them are more specific to their target site than other restriction enzymes. This increases their specificity and reduces their toxicity as they will not target as many sites within a genome. The most studied meganucleases are the LAGLIDADG family. While meganucleases are still quite susceptible to off-target binding, which makes them less attractive than other gene editing tools, their smaller size still makes them attractive particularly for viral vectorization perspectives.[65][53]
Zinc-finger nucleases (ZFNs), used for the first time in 1996, are typically created through the fusion of Zinc-finger domains and the FokI nuclease domain. ZFNs have thus the ability to cleave DNA at target sites.[53] By engineering the zinc finger domain to target a specific site within the genome, it is possible to edit the genomic sequence at the desired location.[65][66][53] ZFNs have a greater specificity, but still hold the potential to bind to non-specific sequences.. While a certain amount of off-target cleavage is acceptable for creating transgenic model organisms, they might not be optimal for all human gene therapy treatments.[65]
TALEN and CRISPR
Access to the code governing the DNA recognition by transcription activator-like effectors (TALE) in 2009 opened the way to the development of a new class of efficient TAL-based gene editing tools. TALE, proteins secreted by the Xanthomonas plant pathogen, bind with great specificity to genes within the plant host and initiate transcription of the genes helping infection. Engineering TALE by fusing the DNA binding core to the FokI nuclease catalytic domain allowed creation of a new tool of designer nucleases, the TALE nuclease (TALEN).[67] They have one of the greatest specificities of all the current engineered nucleases. Due to the presence of repeat sequences, they are difficult to construct through standard molecular biology procedure and rely on more complicated method of such as Golden gate cloning.[62]
In 2011, another major breakthrough technology was developed based on CRISPR/Cas (clustered regularly interspaced short palindromic repeat / CRISPR associated protein) systems that function as an adaptive immune system in bacteria and archaea. The CRISPR/Cas system allows bacteria and archaea to fight against invading viruses by cleaving viral DNA and inserting pieces of that DNA into their own genome. The organism then transcribes this DNA into RNA and combines this RNA with Cas9 proteins to make double-stranded breaks in the invading viral DNA. The RNA serves as a guide RNA to direct the Cas9 enzyme to the correct spot in the virus DNA. By pairing Cas proteins with a designed guide RNA CRISPR/Cas9 can be used to induce double-stranded breaks at specific points within DNA sequences. The break gets repaired by cellular DNA repair enzymes, creating a small insertion/deletion type mutation in most cases. Targeted DNA repair is possible by providing a donor DNA template that represents the desired change and that is (sometimes) used for double-strand break repair by homologous recombination. It was later demonstrated that CRISPR/Cas9 can edit human cells in a dish. Although the early generation lacks the specificity of TALEN, the major advantage of this technology is the simplicity of the design. It also allows multiple sites to be targeted simultaneously, allowing the editing of multiple genes at once. CRISPR/Cpf1 is a more recently discovered system that requires a different guide RNA to create particular double-stranded breaks (leaves overhangs when cleaving the DNA) when compared to CRISPR/Cas9.[62]
CRISPR/Cas9 is efficient at gene disruption. The creation of HIV-resistant babies by Chinese researcher He Jiankui is perhaps the most famous example of gene disruption using this method.[68] It is far less effective at gene correction. Methods of base editing are under development in which a “nuclease-dead” Cas 9 endonuclease or a related enzyme is used for gene targeting while a linked deaminase enzyme makes a targeted base change in the DNA.[69] The most recent refinement of CRISPR-Cas9 is called Prime Editing. This method links a reverse transcriptase to an RNA-guided engineered nuclease that only makes single-strand cuts but no double-strand breaks. It replaces the portion of DNA next to the cut by the successive action of nuclease and reverse transcriptase, introducing the desired change from an RNA template.[70]
See also
- List of genetic engineering software: software to code the genetic modifications
References
- Root C (2007). Domestication. Greenwood Publishing Groups. ISBN 9780313339875.
- Kingsbury N (15 October 2009). Hybrid: The History and Science of Plant Breeding. University of Chicago Press. ISBN 978-0-226-43705-7.
- Hartl DL, Orel V (June 1992). "What did Gregor Mendel think he discovered?". Genetics. 131 (2): 245–53. PMC 1205000. PMID 1644269.
- Roberts RJ (April 2005). "How restriction enzymes became the workhorses of molecular biology". Proceedings of the National Academy of Sciences of the United States of America. 102 (17): 5905–8. Bibcode:2005PNAS..102.5905R. doi:10.1073/pnas.0500923102. PMC 1087929. PMID 15840723.
- Weiss B, Richardson CC (April 1967). "Enzymatic breakage and joining of deoxyribonucleic acid, I. Repair of single-strand breaks in DNA by an enzyme system from Escherichia coli infected with T4 bacteriophage". Proceedings of the National Academy of Sciences of the United States of America. 57 (4): 1021–8. Bibcode:1967PNAS...57.1021W. doi:10.1073/pnas.57.4.1021. PMC 224649. PMID 5340583.
- Lederberg J (October 1952). "Cell genetics and hereditary symbiosis". Physiological Reviews. 32 (4): 403–30. CiteSeerX 10.1.1.458.985. doi:10.1152/physrev.1952.32.4.403. PMID 13003535.
- Mandel M, Higa A (October 1970). "Calcium-dependent bacteriophage DNA infection". Journal of Molecular Biology. 53 (1): 159–62. doi:10.1016/0022-2836(70)90051-3. PMID 4922220.
- Wirth R, Friesenegger A, Fiedler S (March 1989). "Transformation of various species of gram-negative bacteria belonging to 11 different genera by electroporation". Molecular & General Genetics. 216 (1): 175–7. doi:10.1007/BF00332248. PMID 2659971.
- Nester, Eugene. "Agrobacterium: The Natural Genetic Engineer (100 Years Later)". Archived from the original on 19 October 2012. Retrieved 14 January 2011.
- Zambryski P, Joos H, Genetello C, Leemans J, Montagu MV, Schell J (1983). "Ti plasmid vector for the introduction of DNA into plant cells without alteration of their normal regeneration capacity". The EMBO Journal. 2 (12): 2143–50. doi:10.1002/j.1460-2075.1983.tb01715.x. PMC 555426. PMID 16453482.
- "Rediscovering Biology - Online Textbook: Unit 13 Genetically Modified Organisms". www.learner.org. Retrieved 2017-08-18.
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002). "Studying Gene Expression and Function". Molecular Biology of the Cell. New York: Garland Science.
- Griffiths AJ, Miller JH, Suzuki DT, Lewontin RC, Gelbart WM (2000). "Mutant types". An Introduction to Genetic Analysis (7th ed.).
- Koh H, Kwon S, Thomson M (2015-08-26). Current Technologies in Plant Molecular Breeding: A Guide Book of Plant Molecular Breeding for Researchers. Springer. p. 242. ISBN 9789401799966.
- Roh JY, Choi JY, Li MS, Jin BR, Je YH (April 2007). "Bacillus thuringiensis as a specific, safe, and effective tool for insect pest control". Journal of Microbiology and Biotechnology. 17 (4): 547–59. PMID 18051264.
- Adler J (May 2011). "The growing menace from superweeds". Scientific American. 304 (5): 74–9. Bibcode:2011SciAm.304e..74A. doi:10.1038/scientificamerican0511-74. PMID 21595408.
- Food and Agricultural Organisation of the United Nations. "The process of genetic modification".
- Nicholl ST (29 May 2008). An Introduction to Genetic Engineering. Cambridge University Press. ISBN 978-1-139-47178-7.
- Michels CA (10 June 2002). Genetic Techniques for Biological Research: A Case Study Approach. John Wiley & Sons. pp. 85–88. ISBN 978-0-471-89919-8.
- Alberts B, et al. (2002). "8". Isolating, Cloning, and Sequencing DNA (4th ed.). New York: Garland Science.
- Kaufman RI, Nixon BT (July 1996). "Use of PCR to isolate genes encoding sigma54-dependent activators from diverse bacteria". Journal of Bacteriology. 178 (13): 3967–70. doi:10.1128/jb.178.13.3967-3970.1996. PMC 232662. PMID 8682806.
- Liang J, Luo Y, Zhao H (2011). "Synthetic biology: putting synthesis into biology". Wiley Interdisciplinary Reviews: Systems Biology and Medicine. 3 (1): 7–20. doi:10.1002/wsbm.104. PMC 3057768. PMID 21064036.
- "GeneArt Gene Synthesis". www.thermofisher.com. Retrieved 2017-11-09.
- Berg P, Mertz JE (January 2010). "Personal reflections on the origins and emergence of recombinant DNA technology". Genetics. 184 (1): 9–17. doi:10.1534/genetics.109.112144. PMC 2815933. PMID 20061565.
- Nayerossadat N, Maedeh T, Ali PA (6 July 2012). "Viral and nonviral delivery systems for gene delivery". Advanced Biomedical Research. 1: 27. doi:10.4103/2277-9175.98152. PMC 3507026. PMID 23210086.
- Chen I, Dubnau D (March 2004). "DNA uptake during bacterial transformation". Nature Reviews. Microbiology. 2 (3): 241–9. doi:10.1038/nrmicro844. PMID 15083159.
- National Research Council (US) Committee on Identifying and Assessing Unintended Effects of Genetically Engineered Foods on Human Health (2004-01-01). Methods and Mechanisms for Genetic Manipulation of Plants, Animals, and Microorganisms. National Academies Press (US).
- Gelvin SB (March 2003). "Agrobacterium-mediated plant transformation: the biology behind the "gene-jockeying" tool". Microbiology and Molecular Biology Reviews. 67 (1): 16–37, table of contents. doi:10.1128/MMBR.67.1.16-37.2003. PMC 150518. PMID 12626681.
- Francis KE, Spiker S (February 2005). "Identification of Arabidopsis thaliana transformants without selection reveals a high occurrence of silenced T-DNA integrations". The Plant Journal. 41 (3): 464–77. doi:10.1111/j.1365-313x.2004.02312.x. PMID 15659104.
- Schell J, Van Montagu M (1977). "The Ti-Plasmid of Agrobacterium Tumefaciens, A Natural Vector for the Introduction of NIF Genes in Plants?". In Hollaender A, Burris RH, Day PR, Hardy RW, Helinski DR, Lamborg MR, Owens L, Valentine RC (eds.). Genetic Engineering for Nitrogen Fixation. Basic Life Sciences. 9. pp. 159–79. doi:10.1007/978-1-4684-0880-5_12. ISBN 978-1-4684-0882-9. PMID 336023.
- Joos H, Timmerman B, Montagu MV, Schell J (1983). "Genetic analysis of transfer and stabilization of Agrobacterium DNA in plant cells". The EMBO Journal. 2 (12): 2151–60. doi:10.1002/j.1460-2075.1983.tb01716.x. PMC 555427. PMID 16453483.
- Thomson JA. "Genetic Engineering of Plants" (PDF). Biotechnology. 3. Retrieved 17 July 2016.
- Leuzinger K, Dent M, Hurtado J, Stahnke J, Lai H, Zhou X, Chen Q (July 2013). "Efficient agroinfiltration of plants for high-level transient expression of recombinant proteins". Journal of Visualized Experiments. 77 (77). doi:10.3791/50521. PMC 3846102. PMID 23913006.
- Head G, Hull RH, Tzotzos GT (2009). Genetically Modified Plants: Assessing Safety and Managing Risk. London: Academic Pr. p. 244. ISBN 978-0-12-374106-6.
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2002). Molecular Biology of the Cell. New York: Garland Science. p. G:35. ISBN 978-0-8153-4072-0.
- Kamimura K, Suda T, Zhang G, Liu D (October 2011). "Advances in Gene Delivery Systems". Pharmaceutical Medicine. 25 (5): 293–306. doi:10.1007/bf03256872. PMC 3245684. PMID 22200988.
- Pack DW, Hoffman AS, Pun S, Stayton PS (July 2005). "Design and development of polymers for gene delivery". Nature Reviews. Drug Discovery. 4 (7): 581–93. doi:10.1038/nrd1775. PMID 16052241.
- "Lecture 8 genetic engineering of animal cells". www.slideshare.net. 2012-01-25. Retrieved 2018-07-18.
- Biocyclopedia.com. "Gene transfer (transfection) methods in animals | Genetic Engineering and Biotechnology Gene Transfer Methods and Transgenic Organisms | Genetics, Biotechnology, Molecular Biology, Botany | Biocyclopedia.com". biocyclopedia.com. Retrieved 2018-07-18.
- Kim, Tae Kyung; Eberwine, James H. (August 2010). "Mammalian cell transfection: the present and the future". Analytical and Bioanalytical Chemistry. 397 (8): 3173–3178. doi:10.1007/s00216-010-3821-6. ISSN 1618-2642. PMC 2911531. PMID 20549496.
- Mullin A. "Pronuclear Injection". Tulane University.
- "Embryonic Stem Cell-Mediated Gene Transfer - transgenicanimals". sites.google.com. Retrieved 2018-07-19.
- Transduction, Genetic at the US National Library of Medicine Medical Subject Headings (MeSH)
- "Stemcells Redirection". stemcells.nih.gov.
- Tuomela M, Stanescu I, Krohn K (October 2005). "Validation overview of bio-analytical methods". Gene Therapy. 12 Suppl 1 (S1): S131–8. doi:10.1038/sj.gt.3302627. PMID 16231045.
- Narayanaswamy S (1994). Plant Cell and Tissue Culture. Tata McGraw-Hill Education. pp. vi. ISBN 9780074602775.
- Walter, Peter; Roberts, Keith; Raff, Martin; Lewis, Julian; Johnson, Alexander; Alberts, Bruce (2002). "Studying Gene Expression and Function". Molecular Biology of the Cell. 4th Edition.
- Hohn B, Levy AA, Puchta H (April 2001). "Elimination of selection markers from transgenic plants". Current Opinion in Biotechnology. 12 (2): 139–43. doi:10.1016/S0958-1669(00)00188-9. PMID 11287227.
- Setlow JK (2002-10-31). Genetic Engineering: Principles and Methods. Springer Science & Business Media. p. 109. ISBN 9780306472800.
- Deepak S, Kottapalli K, Rakwal R, Oros G, Rangappa K, Iwahashi H, Masuo Y, Agrawal G (June 2007). "Real-Time PCR: Revolutionizing Detection and Expression Analysis of Genes". Current Genomics. 8 (4): 234–51. doi:10.2174/138920207781386960. PMC 2430684. PMID 18645596.
- Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J (2000). "Chapter 8.5: Gene Replacement and Transgenic Animals: DNA Is Transferred into Eukaryotic Cells in Various Ways". Molecular Cell Biology (4th ed.). W. H. Freeman and Company. ISBN 978-0-7167-3136-8.
- Rocha-Martins, Maurício; Cavalheiro, Gabriel R.; Matos-Rodrigues, Gabriel E.; Martins, Rodrigo a. P.; Rocha-Martins, Maurício; Cavalheiro, Gabriel R.; Matos-Rodrigues, Gabriel E.; Martins, Rodrigo a. P. (August 2015). "From Gene Targeting to Genome Editing: Transgenic animals applications and beyond". Anais da Academia Brasileira de Ciências. 87 (2): 1323–1348. doi:10.1590/0001-3765201520140710. ISSN 0001-3765. PMID 26397828.
- Carroll D (August 2011). "Genome engineering with zinc-finger nucleases". Genetics. 188 (4): 773–82. doi:10.1534/genetics.111.131433. PMC 3176093. PMID 21828278.
- Grizot S, Smith J, Daboussi F, Prieto J, Redondo P, Merino N, Villate M, Thomas S, Lemaire L, Montoya G, Blanco FJ, Pâques F, Duchateau P (September 2009). "Efficient targeting of a SCID gene by an engineered single-chain homing endonuclease". Nucleic Acids Research. 37 (16): 5405–19. doi:10.1093/nar/gkp548. PMC 2760784. PMID 19584299.
- Gao H, Smith J, Yang M, Jones S, Djukanovic V, Nicholson MG, West A, Bidney D, Falco SC, Jantz D, Lyznik LA (January 2010). "Heritable targeted mutagenesis in maize using a designed endonuclease". The Plant Journal. 61 (1): 176–87. doi:10.1111/j.1365-313X.2009.04041.x. PMID 19811621.
- Townsend JA, Wright DA, Winfrey RJ, Fu F, Maeder ML, Joung JK, Voytas DF (May 2009). "High-frequency modification of plant genes using engineered zinc-finger nucleases". Nature. 459 (7245): 442–5. Bibcode:2009Natur.459..442T. doi:10.1038/nature07845. PMC 2743854. PMID 19404258.
- Shukla VK, Doyon Y, Miller JC, DeKelver RC, Moehle EA, Worden SE, Mitchell JC, Arnold NL, Gopalan S, Meng X, Choi VM, Rock JM, Wu YY, Katibah GE, Zhifang G, McCaskill D, Simpson MA, Blakeslee B, Greenwalt SA, Butler HJ, Hinkley SJ, Zhang L, Rebar EJ, Gregory PD, Urnov FD (May 2009). "Precise genome modification in the crop species Zea mays using zinc-finger nucleases". Nature. 459 (7245): 437–41. Bibcode:2009Natur.459..437S. doi:10.1038/nature07992. PMID 19404259.
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF (October 2010). "Targeting DNA double-strand breaks with TAL effector nucleases". Genetics. 186 (2): 757–61. doi:10.1534/genetics.110.120717. PMC 2942870. PMID 20660643.
- Li T, Huang S, Jiang WZ, Wright D, Spalding MH, Weeks DP, Yang B (January 2011). "TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain". Nucleic Acids Research. 39 (1): 359–72. doi:10.1093/nar/gkq704. PMC 3017587. PMID 20699274.
- Esvelt KM, Wang HH (2013). "Genome-scale engineering for systems and synthetic biology". Molecular Systems Biology. 9: 641. doi:10.1038/msb.2012.66. PMC 3564264. PMID 23340847.
- Tan WS, Carlson DF, Walton MW, Fahrenkrug SC, Hackett PB (2012). "Precision editing of large animal genomes". Advances in Genetics Volume 80. Advances in Genetics. 80. pp. 37–97. doi:10.1016/B978-0-12-404742-6.00002-8. ISBN 9780124047426. PMC 3683964. PMID 23084873.
- Malzahn, Aimee; Lowder, Levi; Qi, Yiping (2017). "Plant genome editing with TALEN and CRISPR". Cell & Bioscience. 7: 21. doi:10.1186/s13578-017-0148-4. PMC 5404292. PMID 28451378.
- Boissel S, Jarjour J, Astrakhan A, Adey A, Gouble A, Duchateau P, Shendure J, Stoddard BL, Certo MT, Baker D, Scharenberg AM (February 2014). "megaTALs: a rare-cleaving nuclease architecture for therapeutic genome engineering". Nucleic Acids Research. 42 (4): 2591–601. doi:10.1093/nar/gkt1224. PMC 3936731. PMID 24285304.
- Choulika, A.; Perrin, A.; Dujon, B.; Nicolas, J. F. (1994). "The yeast I-Sce I meganuclease induces site-directed chromosomal recombination in mammalian cells". Comptes Rendus de l'Académie des Sciences, Série III. 317 (11): 1013–9. PMID 7882137.
- Silva G, Poirot L, Galetto R, Smith J, Montoya G, Duchateau P, Pâques F (February 2011). "Meganucleases and other tools for targeted genome engineering: perspectives and challenges for gene therapy". Current Gene Therapy. 11 (1): 11–27. doi:10.2174/156652311794520111. PMC 3267165. PMID 21182466.
- Silva GH, Belfort M, Wende W, Pingoud A (August 2006). "From monomeric to homodimeric endonucleases and back: engineering novel specificity of LAGLIDADG enzymes". Journal of Molecular Biology. 361 (4): 744–54. doi:10.1016/j.jmb.2006.06.063. PMID 16872628.
- Christian, M.; Cermak, T.; Doyle, E. L.; Schmidt, C.; Zhang, F.; Hummel, A.; Bogdanove, A. J.; Voytas, D. F. (2010). "Targeting DNA Double-Strand Breaks with TAL Effector Nucleases". Genetics. 186 (2): 757–761. doi:10.1534/genetics.110.120717. PMC 2942870. PMID 20660643.
- Cyranoski, D. (2019). "What's next for CRISPR babies?". Nature. 566: 440–442. doi:10.1038/d41586-019-00673-1. PMID 30809070.
- Rees, H.; Liu, D. R. (2018). "Base editing: precision chemistry on the genome and transcriptome of living cells". Nature Reviews Genetics. 19 (12): 770–788. doi:10.1038/s41576-018-0059-1. PMC 6535181. PMID 30323312.
- Anzalone, A. V.; Randolph, P.B.; Davis, J. R. (2019). "Search-and-replace genome editing without double strand breaks or donor DNA". Nature. 576 (7785): 149–157. Bibcode:2019Natur.576..149A. doi:10.1038/s41586-019-1711-4. PMC 6907074. PMID 31634902.