Duarte galactosemia
Duarte galactosemia is an inherited condition associated with diminished ability to metabolize galactose due to a partial deficiency of the enzyme galactose-1-phosphate uridylyltransferase.[1] DG differs from classic galactosemia in that patients with Duarte galactosemia have partial GALT deficiency whereas patients with classic galactosemia have complete, or almost complete, GALT deficiency.[2] Duarte galactosemia (DG) is much more common than classic galactosemia, and is estimated to affect close to one in 4,000 infants born in the United States. Historically, most healthcare professionals have considered DG to be clinically mild [1] based on pilot studies and anecdotal experience, and in 2019 a large study confirmed that children with DG are not at increased risk for developmental problems relative to children who do not have DG.[3] Due to regional variations in newborn screening (NBS) protocols, some infants with DG are identified by NBS but others are not.[4]
| Duarte galactosemia | |
|---|---|
| Other names | Duarte variant galactosemia, DG, or Biochemical variant galactosemia) |
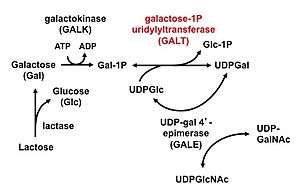 | |
| Leloir metabolic pathway: Galactose-1Puridylyltransferase (GALT, red font) is the middle enzyme in the Leloir pathway of galactose metabolism. | |
Symptoms
Infants with DG often show biochemical differences from infants who do not have DG, especially if exposed to milk, but may not show any acute or developmental symptoms. Specifically, when exposed to high levels of dietary galactose, a sugar abundant in breast milk, milk formula, and most dairy products,[5] infants with DG may show elevated levels of galactose and galactose metabolites such as galactose-1-phosphate (Gal-1P) and galactitol in blood and urine, respectively. Like many infants who do not have DG, some infants with DG may also show acute symptoms of milk sensitivity, such as Jaundice or vomiting, after exposure to milk but these symptoms may reflect sensitivity of the child to components of milk other than galactose, and typically resolve quickly when the baby is switched to a non-dairy diet, such as soy formula.[1] A large study of developmental outcomes in 6- to 12-year-old children with Duarte galactosemia published in 2019[3] demonstrated that children with DG do not show increased prevalence of developmental problems relative to children who do not have DG. This suggests that if a child with DG does show developmental problems, other possible causes should be explored.[1]
Inheritance
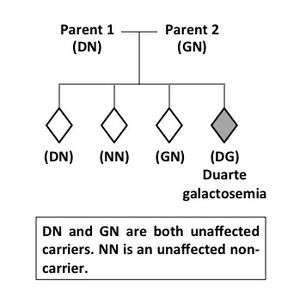
Duarte galactosemia is inherited as a Mendelian autosomal recessive trait. A child with DG carries two different types of GALT alleles, one inherited from each parent. One of these GALT alleles, the G allele, carries a mutation that severely inhibits the function of the encoded GALT enzyme. The other GALT allele, called the D or D2 allele, carries mutations that partially compromise the expression and change some biochemical properties of the encoded GALT enzyme. Together, the G and D alleles only produce about 25% of the normal level of GALT enzyme activity found in a person with two normal (N) GALT alleles.[1] Both parents of a child with DG are considered carriers for GALT variant alleles. One parent carries the G allele and the other carries the D allele. The genotypes of these parents would be written GN and DN, respectively. Without follow-up testing of the parents, it is not possible to know which parent contributed which GALT allele to a child with DG. Like other autosomal recessive conditions, the recurrence risk for DG is 1 in 4, meaning that for each successive child born to parents who already have a child with DG there is a 1 in 4 chance the new baby will also have DG (Figure 2). In rare cases, one parent may actually have DG, while the other parent is a carrier for a G allele (GN). For these couples, there is a 1 in 4 recurrence risk for DG and also a 1 in 4 risk with each pregnancy that the new baby will have classic galactosemia (GG). In extremely rare cases a GALT gene mutation may arise de novo, so that only one parent is a carrier; however, only one case of this has been reported in the literature for galactosemia.[6]
Diagnosis
Infants with DG are generally diagnosed in follow-up to a positive newborn screening (NBS) result for galactosemia. Specifically, dried blood spots collected for NBS from infants with DG may show low (but generally non-zero) GALT enzyme activity, elevated galactose metabolite levels, or both. DG can also be identified by genetic testing.[1]
Of note, not all NBS tests for galactosemia are designed to detect DG so infants with DG born in one jurisdiction may be detected while those born in another may not.[4] For example, all states in the US screen for classic galactosemia in their NBS panel, but some states have lower GALT enzyme activity cut-off levels than others. NBS in states with a low GALT cut off level still detect classic galactosemia, but are likely to miss many infants with DG. In those states, a normal NBS result for galactosemia may not be informative about an infant's DG status.
Most infants with DG who are flagged by a positive NBS result for galactosemia have their diagnosis confirmed in a follow-up evaluation. The differential diagnosis for a positive newborn screening result for galactosemia, especially if based on galactose metabolite levels, includes: classic galactosemia, clinical variant galactosemia, DG, GALE (epimerase) deficiency, GALK (galactokinase) deficiency, or a false positive result.[1] There are also other rare conditions, such as portosystemic venous shunting and hepatic arteriovenous malformations, or Fanconi-Bickel Syndrome (GSDXI) that can lead to elevated blood galactose or urinary galactitol, potentially triggering an initial suspicion of galactosemia.[1][7] If the NBS result is based only on GALT activity and not on metabolite levels then the differential diagnosis would include classic galactosemia, clinical variant galactosemia, DG, and false positive.
Management
Historically, there has been no broadly accepted standard of care for infants with DG.[8] At present, some healthcare providers recommend partial to complete restriction of milk and other high galactose foods for infants with DG; others do not. Because children with DG develop increased tolerance for dietary galactose as they grow, few healthcare providers recommend dietary restriction of galactose beyond early childhood. A revised perspective on clinical care for infants with Duarte galactosemia was published in 2019.[9]
The rationale for NOT restricting milk exposure of infants with DG: Healthcare providers who do not recommend dietary restriction of milk for infants with DG generally consider DG to be of no clinical significance—meaning most infants and children with DG seem to be doing clinically well. A large study reported in 2019[3] supported this conclusion. Further, these providers may be opposed to interrupting or reducing breastfeeding when there is no clear evidence it is contraindicated. These providers may argue that the recognized health benefits of breastfeeding outweigh the potential risks of as yet unknown negative effects of continued milk exposure for these infants.[8] For infants with DG who continue to drink milk, some doctors recommend that blood galactose-1-phosphate (Gal-1P) or urinary galactitol be rechecked by age 12 months to ensure that these metabolite levels are normalizing.[1]
The rationale FOR restricting milk exposure of infants with DG: Healthcare providers who recommend partial or complete dietary restriction of milk for infants with DG generally cite concern about the unknown long-term consequences of abnormally elevated galactose metabolites in a young child's blood and tissues. Infants with DG who continue to drink milk accumulate the same set of abnormal galactose metabolites seen in babies with classic galactosemia – e.g. galactose, Gal-1P, galactonate, and galactitol[10] – but to a lesser extent. While it remains unclear whether any of these metabolites contribute to the long-term developmental complications experienced by so many older children with classic galactosemia, the theoretical possibility that they might cause problems in children with DG serves to motivate some healthcare providers to recommend dietary galactose restriction for infants with DG. Switching an infant with DG from milk or milk formula (high galactose) to a low-galactose formula rapidly normalizes their galactose metabolites. This approach is considered potentially preventative rather than responsive to symptoms.[8] Of course, if a baby with DG, like any other baby, shows acute signs of milk sensitivity then switching the baby to a non-dairy formula would be responsive to those acute symptoms.
If a baby with DG is switched to a low-galactose diet due to concern about elevated galactose metabolites, the healthcare provider may recommend a galactose challenge to re-evaluate galactose tolerance before the restrictive diet is discontinued. Most infants or young children with DG who are followed by a metabolic specialist are discharged from follow up after a successful galactose challenge.
What is a galactose challenge? The goal of a galactose challenge is to learn whether a child is able to metabolize dietary galactose sufficiently to prevent the abnormal accumulation of galactose metabolites, generally measured as Gal-1P in the blood or galactitol in the urine.[1] For infants with DG who showed elevated galactose metabolites at diagnosis, this test can be used to see if the child's ability to process galactose has improved.
For example, to test galactose metabolism, a baseline Gal-1P level is measured while the child is on a galactose-restricted diet. If the level is within the normal range (e.g. <1.0 mg/dL), the parent/guardian is advised to challenge their child with dietary galactose—meaning feed the child a diet that includes normal levels of milk and dairy for 2–4 weeks. Immediately after that time, another blood sample is collected and analyzed for Gal-1P level. If this second result is still in the normal range, the child is said to have passed their galactose challenge, and dietary galactose restrictions are typically relaxed or discontinued. If the second test shows elevated Gal-1P levels, the parent/guardian may be advised to resume galactose restriction for the child, and the challenge may be repeated after a few months.[1]
Prognosis
Until recently, very little was known about outcomes in DG after early childhood. This was because many infants with DG were born in states where they were not diagnosed by NBS, and of those who were diagnosed, most were discharged from metabolic follow-up as toddlers. It was therefore unclear whether older children with DG were at increased risk for long-term developmental problems, and also unclear whether long-term developmental outcomes in DG might be modified by exposure to milk in the first year of life.[1] Of note, premature ovarian insufficiency, a common outcome among girls and women with classic galactosemia, was checked by hormone studies of girls with DG and demonstrated not to be a problem.[11]
Prior Research Concerning Developmental Outcomes of Children with DG: Four studies of developmental outcomes of children with DG have been published.
- The first[12] report published in 2008 was a pilot study that looked at biochemical markers and developmental outcomes in a group of 28 toddlers and young children with DG, some of whom had drunk milk through infancy and some of whom had drunk low-galactose formula. The authors found that galactose metabolites were significantly elevated in the infants drinking milk over those drinking low-galactose formula.[10] However, all of the children scored within the normal range on standardized tests of child development.
- The second study[13] published in 2009 looked at 3-to-10 year-olds living in the greater Atlanta area and asked whether children diagnosed as newborns with DG in this group were more likely than the general population to receive special educational services in school. The answer was yes. Specifically, 5 of the 59 children with DG in this group had received special educational services for speech/language; this proportion was higher than that reported for the local population.
- The third report, published in 2015, was a very small pilot study involving direct assessments of 15 children, all ages 6–11 years old; 15 had DG and 5 did not.[14] Children in the DG group showed slower auditory processing than did the control group. The DG group also showed some slight differences in auditory memory, receptive language/ listening skills, social-emotional functioning, balance, and fine motor coordination.
- The final report addressing developmental outcomes of children with Duarte galactosemia [3] published in 2019, looked at 350 children, ages 6–12 years old: 206 with DG and 144 controls. Of the children with DG, about 40% drank milk as infants and about 60% drank low-galactose formula. The researchers conducted direct assessments in 5 developmental domains for all children yielding 73 outcome scores that were tested for possible association with DG. The “top 10” outcomes were also tested for possible association with milk exposure among study participants with DG. The results clearly demonstrated no significant association of scores for any of the developmental outcomes tested with DG status. The results also demonstrated no significant association of scores for outcomes tested with dietary exposure to milk in infancy among the children with DG in the study. To the limits of the study, these results demonstrate that children with DG are not at increased risk for developmental problems, and that milk exposure in infancy does not lead to developmental problems later in childhood.
Epidemiology
The prevalence of DG in the United States (US) can only be estimated because there is no true population surveillance for this condition. Differences in NBS methods result in very different detection rates in different states. For example, in some US states, DG is detected by NBS in up to 1 in 3500 infants screened, while in other states it is essentially not detected.[4] DG prevalence in the US population is estimated to be approximately 1 in 4,000,[12] which is more than 10 times the prevalence of classic galactosemia.[1][2] Of note, because of different allele frequencies for the G and D2 GALT alleles in different human populations,[15] DG is found predominantly among infants of European ancestry and only very rarely among infants of African or Asian ancestry.
References
- Fridovich-Keil, J., et al., Duarte Variant Galactosemia, in GeneReviews, R. Pagon, et al., Editors. 2014, University of Washington, Seattle. Review. PMID 25473725
- Berry, G., Classic Galactosemia and Clinical Variant Galactosemia, in GeneReviews, R. Pagon, et al., Editors. 2014, University of Washington, Seattle. Review.
- Carlock, G., Fischer, S. T., Lynch, M. E., Potter, N. L., Coles, C. D., Epstein, M. P., ... & Wilson, E., Developmental outcomes in Duarte galactosemia. 2018 Pediatrics, doi: 10.1542/peds.2018-2516. PMID 30593450
- Pyhtila, B.M., et al., Newborn screening for galactosemia in the United States: looking back, looking around, and looking ahead. JIMD Rep, 2015. 15: p. 79-93. Review. PMID 24718839
- Van Calcar SC, Bernstein LE, Rohr FJ, Scaman CH, Yannicelli S, Berry GT. A re-evaluation of life-long severe galactose restriction for the nutrition management of classic galactosemia. Mol Genet Metab. 2014 Jul;112(3):191-7. doi: 10.1016/j.ymgme.2014.04.004. Epub 2014 May 2. Review. PMID 24857409
- Tran, T.T., et al., A De Novo Variant in Galactose-1-P Uridylyltransferase (GALT) Leading to Classic Galactosemia. JIMD Rep, 2015. 19: p. 1-6
- Nishimura Y, Tajima G, Dwi Bahagia A, Sakamoto A, Ono H, Sakura N, Naito K, Hamakawa M, Yoshii C, Kubota M, Kobayashi K, Saheki T. Differential diagnosis of neonatal mild hypergalactosaemia detected by mass screening: clinical significance of portal vein imaging. J Inherit Metab Dis. 2004;27(1):11-8. PMID 14970742
- Fernhoff, P.M., Duarte galactosemia: how sweet is it? Clin Chem, 2010. 56(7): p. 1045-6.
- McCandless, S. E., 2019. Answering a Question Older Than Most Pediatricians: What to Do About Duarte Variant Galactosemia. Pediatrics, 143(1). doi: 10.1542/peds.2018-3292 PMID 30593448
- Ficicioglu, C., et al., Monitoring of biochemical status in children with Duarte galactosemia: utility of galactose, galactitol, galactonate, and galactose 1-phosphate. Clin Chem, 2010. 56(7): p. 1177-82. PMID 20489133
- Badik, J.R., et al., Ovarian function in Duarte galactosemia. Fertil Steril, 2011. 96(2): p. 469-473 e1. PMID 21719007
- Ficicioglu, C., et al., Duarte (DG) galactosemia: a pilot study of biochemical and neurodevelopmental assessment in children detected by newborn screening. Mol Genet Metab, 2008. 95(4): p. 206-12.
- Powell KK, Van Naarden Braun K, Singh RH, Shapira SK, Olney RS, Yeargin-Allsopp M. Long-term speech and language developmental issues among children with Duarte galactosemia. Genet Med. 2009 Dec;11(12):874-9. doi: 10.1097/GIM.0b013e3181c0c38d. PMID 19904210
- Lynch ME, Potter NL, Coles CD, Fridovich-Keil JL. Developmental Outcomes of School-Age Children with Duarte Galactosemia: A Pilot Study. JIMD Rep. 2015;19:75-84. doi: 10.1007/8904_2014_370. Epub 2015 Feb 15.PMID 25681083
- Carney et al. Origins, distribution and expression of the Duarte-2 (D2) allele of galactose-1-phosphate uridylyltransferase. Hum Mol Genet. 2009 May 1;18(9):1624-32. doi: 10.1093/hmg/ddp080. Epub 2009 Feb 18.
Further reading
- Fernandes, John; Saudubray, Jean-Marie; Berghe, Georges van den (2013-03-14). Inborn Metabolic Diseases: Diagnosis and Treatment. Springer Science & Business Media. ISBN 9783662031476.
- Leonard, Debra G. B. (2007-11-25). Molecular Pathology in Clinical Practice. Springer Science & Business Media. ISBN 9780387332277.
External links
- Duarte Galactosemia Website
- The Galactosemia Foundation website
- Duarte Variant Galactosemia article in GeneReviews
- Duarte Galactosemia Study summary on PCORI.org