D-lysine 5,6-aminomutase
In enzymology, D-lysine 5,6-aminomutase (EC 5.4.3.4) is an enzyme that catalyzes the chemical reaction
- D-lysine 2,5-diaminohexanoate

| D-Lysine 5,6-aminomutase alpha subunit | |||||||||
|---|---|---|---|---|---|---|---|---|---|
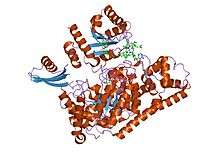 crystal structure of lysine 5,6-aminomutase in complex with plp, cobalamin, and 5'-deoxyadenosine | |||||||||
| Identifiers | |||||||||
| Symbol | Lys-AminoMut_A | ||||||||
| Pfam | PF09043 | ||||||||
| InterPro | IPR015130 | ||||||||
| |||||||||
| D-lysine 5,6-aminomutase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC number | 5.4.3.4 | ||||||||
| CAS number | 9075-70-1 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
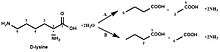
Hence, this enzyme has one substrate, D-lysine, and one product, 2,5-diaminohexanoate.
This enzyme participates in lysine degradation. It employs one cofactor, cobamide.
Background
D-lysine 5,6-aminomutase belongs to the isomerase family of enzymes, specifically intramolecular transferases, which transfers amino groups. Its systematic name is D-2,6-diaminohexanoate 5,6-aminomutase. Other names in common use include D-α-lysine mutase and adenosylcobalamin-dependent D-lysine 5,6-aminomutase, which can be abbreviated as 5,6-LAM.
5,6-LAM is capable of reversibly catalyzing the migration of an amino group from ε-carbon to δ-carbon in both D-lysine and L-β-lysine, and catalyzing the migration of hydrogen atoms from δ-carbon to ε-carbon at the same time.[1] It demonstrates greatest catalytic activity in 20mM Tris•HCl at pH 9.0-9.2.[2]
In the early 1950s, 5,6-LAM was discovered in the amino-acid-fermenting bacteria Clostridium sticklandii, in which lysine undergoes degradation under anaerobic conditions to equimolar amounts of acetate and butyrate.[3]
Later, isotopic studies uncovered two possible pathways. In pathway A, both acetate and butyrate are generated from C2-C3 cleavage of D-lysine. Unlike pathway A, pathway B involves C5-C4 degradation, producing the same products.
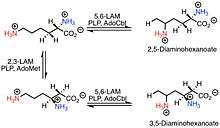
D-lysine 5,6-aminomutase (5,6-LAM) is responsible for the first conversion in pathway B to convert D-α-lysine into 2,5-diaminohexanoate. Unlike other members of the family of aminomutases (like 2,3-LAM), which are peculiar to a single substrate, 5,6-LAM can reversibly catalyze both the reaction of D-lysine to 2,5-diaminohexanoic acid and the reaction of L-β-lysine to 3,5-diaminohexanoic acid.[3][4]
Structure
Subunits
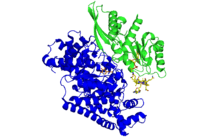
5,6-LAM is an α2β2 tetramer. The structure of the alpha subunit is predominantly a PLP-binding TIM barrel domain, with several additional alpha-helices and beta-strands at the N and C termini. These helices and strands form an intertwined accessory clamp structure that wraps around the sides of the TIM barrel and extends up toward the Ado ligand of the Cbl cofactor, which is the beta subunit providing most of the interactions observed between the protein and the Ado ligand of the Cbl, suggesting that its role is mainly in stabilizing AdoCbl in the precatalytic resting state.[5] The β subunit binds AdoCbl while the PLP directly binds to α subunit. PLP also directly binds to Lys144 of the β subunit to form an internal aldimine. PLP and AdoCbl are separated by a distance of 24Å.[6]
Cofactors
- 5,6-LAM is pyridoxal-5'-phosphate (PLP) dependent. PLP binds to its substrate with an external aldimine linkage. PLP is also important for stabilizing the radical intermediate by captodative stabilization and spin delocalization.[7]
- Catalysis begins with a 5'-deoxyadenosyl radical (Ado-CH2•), and 5'-deoxyadenosylcobalamin (AdoCbl) is an essential cofactor as a hydrogen carrier.[8]
- ATP, a mercaptan, and a divalent metal ion (usually Mg2+) are required to achieve the highest catalytic effect.[4]
Mechanism
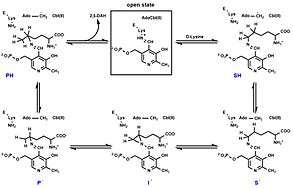
Catalytic cycle
The catalytic cycle starts with Ado-CH2• (5'-deoxyadenosyl radical derived from adenosylcobalamine) abstracting a hydrogen atom from PLP-D-lysine adduct (substrate-related precursor SH) to generate a substrate-related radical (S•), with the radical located at carbon 5 of the lysine residue. The latter undergoes an internal cyclization/addition to the imine nitrogen producing an aziridinecarbinyl radical (I•) — a more thermodynamically stable intermediate with the radical being at a benzylic position. Rearrangement of I• produces a product-related radical (P•), which then participates in the final step of hydrogen transfer from AdoH to afford the PLP-product complex (PH).[9]
Structure-based catalysis
Further understanding of the catalytic mechanism can be derived from the X-ray structure.
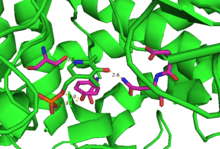
First, an evident conformational change is observed after the substrate is added to the system. With a substrate-free enzyme, the distance between AdoCbl and PLP is about 24 Å. PLP participates in multiple non-covalent interactions with the enzyme with 5,6-LAM presenting an “open” state.
The first step of the catalytic cycle involves the enzyme accepting the substrate by forming an external aldimine with PLP replacing the PLP-Lys144β internal aldimine. With the cleavage of the internal aldimine, the β unit is able to swing towards to the top of the α unit and block the empty site. Therefore, generation of the Ado-CH2• radical leads to a change in the structure of the active domain, bringing the AdoCbl and PLP-substrate complex closer to each other, thus locking the enzyme in a “closed” state. The closed state exists until the radical transfer occurs when the product is released and AdoCbl is reformed. At the same time, the closed state is transformed to the open state again to wait for the next substrate.[10]
Also worth mentioning is the locking mechanism to prevent the radical reaction without the presence of substrate discovered by Catherine Drennan's group. Lys144 of the β subunit is located at a short G-rich loop highly conserved across all 5,6-LAMs, which blocks the AdoCbl from the reaction site. Based on X-ray structure analysis, when the open structure is applied, the axes of the TIM barrel and Rossmann domains are in different directions. With the addition of the substrate, the subunits rearrange to turn the axes into each other to facilitate the catalysis.[11] For example, in wild type 5,6-LAM, the phenol ring of Tyr263α is oriented in a slipped geometry with pyridine ring of PLP, generating a π-π stacking interaction, which is capable of modulating the electron distribution of the high-energetic radical intermediate.[12]
History
Early insights into the mechanism of the catalytic reaction mainly focused on isotopic methods. Both pathways of lysine degradation and the role of 5,6-LAM were discovered in early work by Stadtman's group during 1950s-1960s. In 1971, having a tritiated α-lysine, 2,5-diaminohexanoate, and coenzyme in hand, Colin Morley and T. Stadtman discovered the role of 5'-deoxyadenosylcobalamin (AdoCbl) as a source for hydrogen migration.[8] Recently, much progress has been made toward detecting the intermediates of the reaction, especially towards I•. Based on quantum-mechanical calculations, it was proposed that with 5-fluorolysine[9] as a substitute for D-lysine the 5-FS• species can be captured and analyzed. A similar approach was applied towards PLP modification, when it was modified to 4’-cyanoPLP[13] or PLP-NO.[14] The radical intermediate I• analogue is hypothesized to be easily detected to support the proposed mechanism. Other simulations can also provide some insights into the catalytic reaction.[1]
References
- Sandala GM, Smith DM, Radom L (December 2006). "In search of radical intermediates in the reactions catalyzed by lysine 2,3-aminomutase and lysine 5,6-aminomutase". Journal of the American Chemical Society. 128 (50): 16004–5. doi:10.1021/ja0668421. PMID 17165731.
- Morley CG, Stadtman TC (December 1970). "Studies on the fermentation of D-alpha-lysine. Purification and properties of an adenosine triphosphate regulated B 12-coenzyme-dependent D-alpha-lysine mutase complex from Clostridium sticklandii". Biochemistry. 9 (25): 4890–900. doi:10.1021/bi00827a010. PMID 5480154.
- Stadtman TC, White FH (June 1954). "Tracer studies on ornithine, lysine, and formate metabolism in an amino acid fermenting Clostridium". Journal of Bacteriology. 67 (6): 651–7. PMC 357300. PMID 13174491.
- Stadtman TC, Tsai L (September 1967). "A cobamide coenzyme dependent migration of the epsilon-amino group of D-lysine". Biochemical and Biophysical Research Communications. 28 (6): 920–6. doi:10.1016/0006-291x(67)90067-8. PMID 4229021.
- Berkovitch F, Behshad E, Tang KH, Enns EA, Frey PA, Drennan CL (November 2004). "A locking mechanism preventing radical damage in the absence of substrate, as revealed by the x-ray structure of lysine 5,6-aminomutase". Proceedings of the National Academy of Sciences of the United States of America. 101 (45): 15870–5. doi:10.1073/pnas.0407074101. PMC 528771. PMID 15514022.
- Lo HH, Lin HH, Maity AN, Ke SC (May 2016). "The molecular mechanism of the open-closed protein conformational cycle transitions and coupled substrate binding, activation and product release events in lysine 5,6-aminomutase". Chemical Communications. 52 (38): 6399–402. doi:10.1039/c6cc01888b. PMID 27086547.
- Chen YH, Maity AN, Pan YC, Frey PA, Ke SC (November 2011). "Radical stabilization is crucial in the mechanism of action of lysine 5,6-aminomutase: role of tyrosine-263α as revealed by electron paramagnetic resonance spectroscopy". Journal of the American Chemical Society. 133 (43): 17152–5. doi:10.1021/ja207766c. PMID 21939264.
- Morley CG, Stadtman TC (June 1971). "Studies on the fermentation of p-alpha-lysine. On the hydrogen shift catalyzed by the B 12 coenzyme dependent D-alpha-lysine mutase". Biochemistry. 10 (12): 2325–9. doi:10.1021/bi00788a023. PMID 5114991.
- Maity AN, Ke S (October 2013). "5-Fluorolysine as alternative substrate of lysine 5,6-aminomutase: A computational study". Computational and Theoretical Chemistry. 1022: 1–5. doi:10.1016/j.comptc.2013.08.007.
- Chen Y, Maity AN, Frey PA, Ke S (January 2013). "Mechanism-based Inhibition Reveals Transitions between Two Conformational States in the Action of Lysine 5,6-Aminomutase: A Combination of Electron Paramagnetic Resonance Spectroscopy, Electron Nuclear Double Resonance Spectroscopy, and Density Functional Theory Study". Journal of the American Chemical Society. 135 (2): 788–794. doi:10.1021/ja309603a.
- Berkovitch F, Behshad E, Tang KH, Enns EA, Frey PA, Drennan CL (November 2004). "A locking mechanism preventing radical damage in the absence of substrate, as revealed by the x-ray structure of lysine 5,6-aminomutase". Proceedings of the National Academy of Sciences of the United States of America. 101 (45): 15870–5. doi:10.1073/pnas.0407074101. PMC 528771. PMID 15514022.
- Wetmore SD, Smith DM, Radom L (September 2001). "Enzyme catalysis of 1,2-amino shifts: the cooperative action of B6, B12, and aminomutases". Journal of the American Chemical Society. 123 (36): 8678–89. doi:10.1021/ja010211j. PMID 11535072.
- Maity AN, Ke SC (February 2015). "4'-CyanoPLP presents better prospect for the experimental detection of elusive cyclic intermediate radical in the reaction of lysine 5,6-aminomutase". Biochemical and Biophysical Research Communications. 457 (2): 161–4. doi:10.1016/j.bbrc.2014.12.076. PMID 25542154.
- Maity AN, Lin H, Chiang H, Lo H, Ke S (May 2015). "Reaction of Pyridoxal-5′-phosphate-N-oxide with Lysine 5,6-Aminomutase: Enzyme Flexibility toward Cofactor Analog". ACS Catalysis. 5 (5): 3093–3099. doi:10.1021/acscatal.5b00671.