Crigler–Najjar syndrome
Crigler–Najjar syndrome is a rare inherited disorder affecting the metabolism of bilirubin, a chemical formed from the breakdown of the heme in red blood cells. The disorder results in a form of nonhemolytic jaundice, which results in high levels of unconjugated bilirubin and often leads to brain damage in infants. The disorder is inherited in an autosomal recessive manner.
| Crigler–Najjar syndrome | |
|---|---|
| Other names | CNS |
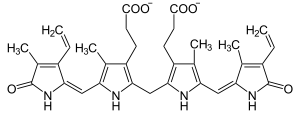 | |
| Bilirubin | |
| Specialty | Pediatrics, hepatology |
This syndrome is divided into types I and II, with the latter sometimes called Arias syndrome. These two types, along with Gilbert's syndrome, Dubin–Johnson syndrome, and Rotor syndrome, make up the five known hereditary defects in bilirubin metabolism. Unlike Gilbert's syndrome, only a few causes of CNS are known.
Cause
It is caused by abnormalities in the gene coding for uridine diphosphoglucuronate glucuronosyltransferase (UGT1A1). UGT1A1 normally catalyzes the conjugation of bilirubin and glucuronic acid within hepatocytes. Conjugated bilirubin is more water soluble and is excreted in bile.
Diagnosis
Type I
This is a very rare disease (estimated at 0.6–1.0 per million live births), and consanguinity increases the risk of this condition (other rare diseases may be present). Inheritance is autosomal recessive.
Intense jaundice appears in the first days of life and persists thereafter. Type 1 is characterised by a serum bilirubin usually above 345 µmol/L [20 mg/dL] (range 310–755 µmol/L [18–44 mg/dL]) (whereas the reference range for total bilirubin is 2–14 μmol/L [0.1–0.8 mg/dL]).
No UDP glucuronosyltransferase 1-A1 expression can be detected in the liver tissue. Hence, there is no response to treatment with phenobarbital,[1] which causes CYP450 enzyme induction. Most patients (type IA) have a mutation in one of the common exons (2 to 5), and have difficulties conjugating several additional substrates (several drugs and xenobiotics). A smaller percentage of patients (type IB) have mutations limited to the bilirubin-specific A1 exon; their conjugation defect is mostly restricted to bilirubin itself.
Before the availability of phototherapy, these children died of kernicterus (bilirubin encephalopathy) or survived until early adulthood with clear neurological impairment. Today, therapy includes
- exchange transfusions in the immediate neonatal period
- 12 hours/day phototherapy
- heme oxygenase inhibitors to reduce transient worsening of hyperbilirubinemia (although the effect decreases over time)
- oral calcium phosphate and carbonate to form complexes with bilirubin in the gut
- liver transplantation before the onset of brain damage and before phototherapy becomes ineffective at later age
Type II
The inheritance patterns of both Crigler–Najjar syndrome types I and II are autosomal recessive.[2]
However, type II differs from type I in a number of different aspects:
- Bilirubin levels are generally below 345 µmol/L [20 mg/dL] (range 100–430 µmol/L [6–24 mg/dL]; thus, overlap may sometimes occur), and some cases are only detected later in life.
- Because of lower serum bilirubin, kernicterus is rare in type II.
- Bile is pigmented, instead of pale in type I or dark as normal, and monoconjugates constitute the largest fraction of bile conjugates.
- UGT1A1 is present at reduced but detectable levels (typically <10% of normal), because of single base pair mutations.
- Therefore, treatment with phenobarbital is effective, generally with a decrease of at least 25% in serum bilirubin. In fact, this can be used, along with these other factors, to differentiate type I and II.
Differential diagnosis
Neonatal jaundice may develop in the presence of sepsis, hypoxia, hypoglycemia, hypothyroidism, hypertrophic pyloric stenosis, galactosemia, fructosemia, etc.
Hyperbilirubinemia of the unconjugated type may be caused by:
- increased production
- hemolysis (e.g., hemolytic disease of the newborn, hereditary spherocytosis, sickle cell disease)
- ineffective erythropoiesis
- massive tissue necrosis or large hematomas
- decreased clearance
- drug-induced
- physiological neonatal jaundice and prematurity
- liver diseases such as advanced hepatitis or cirrhosis
- breast milk jaundice and Lucey–Driscoll syndrome
- Crigler–Najjar syndrome and Gilbert syndrome
In Crigler–Najjar syndrome and Gilbert syndrome, routine liver function tests are normal, and hepatic histology usually is normal, too. No evidence for hemolysis is seen. Drug-induced cases typically regress after discontinuation of the substance. Physiological neonatal jaundice may peak at 85–170 µmol/l and decline to normal adult concentrations within two weeks. Prematurity results in higher levels.
Treatment
Plasmapheresis and phototherapy are used for treatment. Liver transplant is curative.
Research
A San Francisco based company named Audentes Therapeutics is currently investigating the treatment of Crigler-Najjar syndrome with one of their gene replacement therapy products, AT342. Preliminary success has been found in early stages of a phase 1/2 clinical trial.[3]
One 10-year-old girl with Crigler–Najjar syndrome type I was successfully treated by liver cell transplantation.[4]
The homozygous Gunn rat, which lacks the enzyme uridine diphosphate glucuronyltransferase (UDPGT), is an animal model for the study of Crigler–Najjar syndrome. Since only one enzyme is working improperly, gene therapy for Crigler-Najjar is a theoretical option which is being investigated.[5]
Eponym
The condition is named for John Fielding Crigler (1919 – May 13, 2018), an American pediatrician and Victor Assad Najjar (1914–2002), a Lebanese-American pediatrician.[6][7]
References
- Jansen PL (December 1999). "Diagnosis and management of Crigler–Najjar syndrome". European Journal of Pediatrics. 158 (Suppl 2): S89–S94. doi:10.1007/PL00014330. PMID 10603107.
- Crigler Najjar Syndrome. National Organization for Rare Disorders (NORD). 2016; https://rarediseases.org/rare-diseases/crigler-najjar-syndrome/.
- "AT342 – Crigler-Najjar Syndrome – Audentes Therapeutics". www.audentestx.com. Retrieved 2019-03-09.
- Fox IJ, Chowdhury JR, Kaufman SS, Goertzen TC, Chowdhury NR, Warkentin PI, Dorko K, Sauter BV, Strom SC (May 1998). "Treatment of the Crigler–Najjar syndrome type I with hepatocyte transplantation". The New England Journal of Medicine. 338 (20): 1422–6. doi:10.1056/NEJM199805143382004. PMID 9580649.
- Toietta G, Mane VP, Norona WS, Finegold MJ, Ng P, Mcdonagh AF, Beaudet AL, Lee B (March 2005). "Lifelong elimination of hyperbilirubinemia in the Gunn rat with a single injection of helper-dependent adenoviral vector". Proceedings of the National Academy of Sciences of the United States of America. 102 (11): 3930–5. Bibcode:2005PNAS..102.3930T. doi:10.1073/pnas.0500930102. PMC 554836. PMID 15753292.
- Crigler JF Jr, Najjar VA (February 1952). "Congenital familial nonhemolytic jaundice with kernicterus; a new clinical entity". American Journal of Diseases of Children. 83 (2): 259–60. ISSN 0096-8994. PMID 14884759.
- synd/86 at Who Named It?
External links
| Classification | |
|---|---|
| External resources |
- Crigler–Najjar syndrome, type 1 at NIH's Office of Rare Diseases
- Crigler–Najjar syndrome, type 2 at NIH's Office of Rare Diseases