Congenital stationary night blindness
Congenital stationary night blindness (CSNB) is a rare non-progressive retinal disorder. People with CSNB often have difficulty adapting to low light situations due to impaired photoreceptor transmission. These patients may also have reduced visual acuity, myopia, nystagmus, and strabismus. CSNB has two forms -- complete, also known as type-1 (CSNB1), and incomplete, also known as type-2 (CSNB2), which are distinguished by the involvement of different retinal pathways. In CSNB1, downstream neurons called bipolar cells are unable to detect neurotransmission from photoreceptor cells. CSNB1 can be caused by mutations in various genes involved in neurotransmitter detection, including NYX, GRM6, and TRPM1. In CSNB2, the photoreceptors themselves have impaired neurotransmission function; this is caused primarily by mutations in the gene CACNA1F, which encodes a voltage-gated calcium channel important for neurotransmitter release.
| Congenital stationary night blindness | |
|---|---|
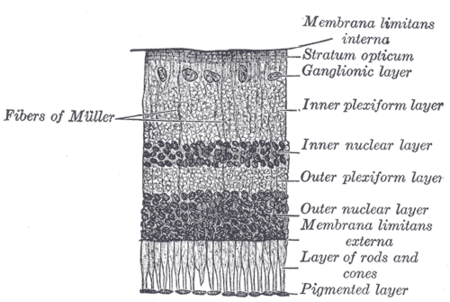 | |
| Malfunction in transmission from the photoreceptors in the outer nuclear layer to bipolar cells in the inner nuclear layer underlies CSNB. | |
| Specialty | Ophthalmology |
Congenital stationary night blindness (CSNB) can be inherited in an X-linked, autosomal dominant, or autosomal recessive pattern, depending on the genes involved.
Symptoms
The X-linked varieties of congenital stationary night blindness (CSNB) can be differentiated from the autosomal forms by the presence of myopia, which is typically absent in the autosomal forms. Patients with CSNB often have impaired night vision, myopia, reduced visual acuity, strabismus and nystagmus. Individuals with the complete form of CSNB (CSNB1) have highly impaired rod sensitivity (reduced ~300x) as well as cone dysfunction. Patients with the incomplete form can present with either myopia or hyperopia.[1]
Cause
CSNB is caused by malfunctions in neurotransmission from rod and cone photoreceptors to bipolar cells in the retina.[2] At this first synapse, information from photoreceptors is divided into two channels: ON and OFF. The ON pathway detects light onset, while the OFF pathway detects light offset.[3] The malfunctions in CSNB1 specifically affect the ON pathway, by hindering the ability of ON-type bipolar cells to detect neurotransmitter released from photoreceptors.[2] Rods, which are responsible for low-light vision, make contacts with ON-type bipolar cells only, while, cones, which are responsible for bright-light vision, make contacts with bipolar cells of both ON an OFF subtypes.[4] Because the low-light sensing rods feed only into the ON pathway, individuals with CSNB1 typically have problems with night vision, while vision in well-lit conditions is spared.[2] In CSNB2, release of neurotransmitter from photoreceptors is impaired, leading to involvement of both ON and OFF pathways.
The electroretinogram (ERG) is an important tool for diagnosing CSNB. The ERG a-wave, which reflects the function of the phototransduction cascade in response to a light flashes, is typically normal in CSNB patients, although in some cases phototransduction is also affected, leading to a reduced a-wave. The ERG b-wave, which primarily reflects the function of ON-bipolar cells, is greatly reduced in CSNB2 cases, and completely absent in CSNB1 cases.[2][5]
Pathophysiology
CSNB1
The complete form of X-linked congenital stationary night blindness, also known as nyctalopia, is caused by mutations in the NYX gene (Nyctalopin on X-chromosome), which encodes a small leucine-rich repeat (LRR) family protein of unknown function.[6][7] This protein consists of an N-terminal signal peptide and 11 LRRs (LRR1-11) flanked by cysteine-rich LRRs (LRRNT and LRRCT). At the C-terminus of the protein there is a putative GPI anchor site. Although the function of NYX is yet to be fully understood, it is believed to be located extracellularly. A naturally occurring deletion of 85 bases in NYX in some mice leads to the "nob" (no b-wave) phenotype, which is highly similar to that seen in CSNB1 patients.[8] NYX is expressed primarily in the rod and cone cells of the retina. There are currently almost 40 known mutations in NYX associated with CSNB1, Table 1., located throughout the protein. As the function of the nyctalopin protein is unknown, these mutations have not been further characterized. However, many of them are predicted to lead to truncated proteins that, presumably, are non-functional.
| Mutation | Position | References | |
|---|---|---|---|
| Nucleotide | Amino acid | ||
| c.?-1_?-61del | 1_20del | Signal sequence | [7] |
| Splicing | Intron 1 | [9] | |
| c.?-63_1443-?del | 21_481del | [7] | |
| c.48_64del | L18RfsX108 | Signal sequence | [9] |
| c.85_108del | R29_A36del | N-terminal LRR | [6] |
| c.G91C | C31S | LRRNT | [7] |
| c.C105A | C35X | LRRNT | [7] |
| c.C169A | P57T | LRRNT | [10] |
| c.C191A | A64E | LRR1 | [10] |
| c.G281C | R94P | LRR2 | [11] |
| c.301_303del | I101del | LRR2 | [7] |
| c.T302C | I101T | LRR2 | [11] |
| c.340_351del | E114_A118del | LRR3 | [7][9] |
| c.G427C | A143P | LRR4 | [7] |
| c.C452T | P151L | LRR4 | [6] |
| c.464_465insAGCGTGCCCGAGCGCCTCCTG | S149_V150dup+P151_L155dup | LRR4 | [6] |
| c.C524G | P175R | LRR5 | [7] |
| c.T551C | L184P | LRR6 | [6] |
| c.556_618delins | H186?fsX260 | LRR6 | [6] |
| c.559_560delinsAA | A187K | LRR6 | [7] |
| c.613_621dup | 205_207dup | LRR7 | [6][7] |
| c.628_629ins | R209_S210insCLR | LRR7 | [6] |
| c.T638A | L213Q | LRR7 | [6] |
| c.A647G | N216S | LRR7 | [6][9] |
| c.T695C | L232P | LRR8 | [6] |
| c.727_738del | 243_246del | LRR8 | [7] |
| c.C792G | N264K | LRR9 | [6] |
| c.T854C | L285P | LRR10 | [6] |
| c.T893C | F298S | LRR10 | [6] |
| c.C895T | Q299X | LRR10 | [9] |
| c.T920C | L307P | LRR11 | [7] |
| c.A935G | N312S | LRR11 | [7] |
| c.T1040C | L347P | LRRCT | [7] |
| c.G1049A | W350X | LRRCT | [6] |
| c.G1109T | G370V | LRRCT | [7] |
| c.1122_1457del | S374RfsX383 | LRRCT | [7][9] |
| c.1306del | L437WfsX559 | C-terminus | [9] |
| LRR: leucine-rich repeat, LRRNT and LRRCT: N- and C-terminal cysteine-rich LRRs. | |||
CSNB2
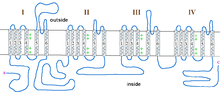
The incomplete form of X-linked congenital stationary night blindness (CSNB2) is caused by mutations in the CACNA1F gene, which encodes the voltage-gated calcium channel CaV1.4 expressed heavily in retina.[12][13] One of the important properties of this channel is that it inactivates at an extremely low rate. This allows it to produce sustained Ca2+ entry upon depolarization. As photoreceptors depolarize in the absence of light, CaV1.4 channels operate to provide sustained neurotransmitter release upon depolarization.[14] This has been demonstrated in CACNA1F mutant mice that have markedly reduced photoreceptor calcium signals.[15] There are currently 55 mutations in CACNA1F located throughout the channel, Table 2 and Figure 1. While most of these mutations result in truncated and, likely, non-functional channels, it is expected that they prevent the ability of light to hyperpolarize photoreceptors. Of the mutations with known functional consequences, 4 produce channels that are either completely non-functional, and two that result in channels which open at far more hyperpolarized potentials than wild-type. This will result in photoreceptors that continue to release neurotransmitter even after light-induced hyperpolarization.
| Mutation | Position | Effect | References | |
|---|---|---|---|---|
| Nucleotide | Amino Acid | |||
| c.C148T | R50X | N-terminus | [16] | |
| c.151_155delAGAAA | R51PfsX115 | N-terminus | [17] | |
| c.T220C | C74R | N-terminus | [17] | |
| c.C244T | R82X | N-terminus | [16][17] | |
| c.466_469delinsGTAGGGGTGCT CCACCCCGTAGGGGTGCTCCACC |
S156VdelPinsGVKHOVGVLH | D1S2-3 | [16][18][19] | |
| Splicing | Intron 4 | [16] | ||
| c.T685C | S229P | D1S4-5 | [17] | |
| c.G781A | G261R | D1-pore | [17] | |
| c.G832T | E278X | D1-pore | [9][20] | |
| c.904insG | R302AfsX314 | D1-pore | [18] | |
| c.951_953delCTT | F318del | D1-pore | [16] | |
| c.G1106A | G369D | D1S6 | Activates ~20mV more negative than wild-type, increases time to peak current and decreases inactivation, increased Ca2+ permeability. | [12][14][16][17][21] |
| c.1218delC | W407GfsX443 | D1-2 | [13][16][20] | |
| c.C1315T | Q439X | D1-2 | [17] | |
| c.G1556A | R519Q | D1-2 | Decreased expression | [12][22] |
| c.C1873T | R625X | D2S4 | [16][17] | |
| c.G2021A | G674D | D2S5 | [14][16][18] | |
| c.C2071T | R691X | D2-pore | [10] | |
| c.T2258G | F753C | D2S6 | [17] | |
| c.T2267C | I756T | D2S6 | Activates ~35mV more negative than wild-type, inactivates more slowly | [23] |
| Splicing | Intron 19 | [17] | ||
| c.T2579C | L860P | D2-3 | [17] | |
| c.C2683T | R895X | D3S1-2 | [9][10][13][16] | |
| Splicing | Intron 22 | [17][18] | ||
| Splicing | Intron 22 | [17] | ||
| c.C2783A | A928D | D3S2-3 | [14][16] | |
| c.C2905T | R969X | D3S4 | [12][17] | |
| c.C2914T | R972X | D3S4 | [20] | |
| Splicing | Intron24 | [16] | ||
| c.C2932T | R978X | D3S4 | [18] | |
| c.3006_3008delCAT | I1003del | D3S4-5 | [16] | |
| c.G3052A | G1018R | D3S5 | [17] | |
| c.3125delG | G1042AfsX1076 | D3-pore | [16] | |
| c.3166insC | L1056PfsX1066 | D3-pore | [12][13][16][17] | |
| c.C3178T | R1060W | D3-pore | [12][17] | |
| c.T3236C | L1079P | D3-pore | Does not open without BayK, activates ~5mV more negative than wild-type | [17][21] |
| c.3672delC | L1225SfsX1266 | D4S2 | [13][16] | |
| c.3691_3702del | G1231_T1234del | D4S2 | [12][17] | |
| c.G3794T | S1265I | D4S3 | [10] | |
| c.C3886A | R1296S | D4S4 | [10] | |
| c.C3895T | R1299X | D4S4 | [13][16][17] | |
| Splicing | Intron 32 | [17] | ||
| c.C4075T | Q1359X | D4-pore | [12][17] | |
| c.T4124A | L1375H | D4-pore | Decreased expression | [12][17][22] |
| Splicing | Intron 35 | [17] | ||
| c.G4353A | W1451X | C-terminus | Non-functional | [13][14][16][21] |
| c.T4495C | C1499R | C-terminus | [17] | |
| c.C4499G | P1500R | C-terminus | [17] | |
| c.T4523C | L1508P | C-terminus | [17] | |
| Splicing | intron 40 | [16] | ||
| c.4581delC | F1528LfsX1535 | C-terminus | [24] | |
| c.A4804T | K1602X | C-terminus | [12][17] | |
| c.C5479T | R1827X | C-terminus | [17] | |
| c.5663delG | S1888TfsX1931 | C-terminus | [16] | |
| c.G5789A | R1930H | C-terminus | [10] | |
Genetics
Only three rhodopsin mutations have been found associated with congenital stationary night blindness (CSNB).[25] Two of these mutations are found in the second transmembrane helix of rhodopsin at Gly-90 and Thr-94. Specifically, these mutations are the Gly90Asp [26] and the Thr94Ile, which has been the most recent one reported.[27] The third mutation is Ala292Glu, and it is located in the seventh transmembrane helix, in proximity to the site of retinal attachment at Lys-296.[28] Mutations associated with CSNB affect amino acid residues near the protonated Schiff base (PSB) linkage. They are associated with changes in conformational stability and the protonated status of the PSB nitrogen.[29]
Footnotes
- Boycott K, Pearce W, Musarella M, Weleber R, Maybaum T, Birch D, Miyake Y, Young R, Bech-Hansen N (1998). "Evidence for genetic heterogeneity in X-linked congenital stationary night blindness". Am J Hum Genet. 62 (4): 865–875. doi:10.1086/301781. PMC 1377021. PMID 9529339.
- Zeitz C, Robson AG, Audo I (2015). "Congenital stationary night blindness: an analysis and update of genotype-phenotype correlations and pathogenic mechanisms". Prog Retin Eye Res. 45: 58–110. doi:10.1016/j.preteyeres.2014.09.001. PMID 25307992.
- Euler T, Haverkamp S, Schubert T, Baden T (2014). "Retinal bipolar cells: elementary building blocks of vision". Nat Rev Neurosci. 15 (8): 507–519. doi:10.1038/nrn3783. PMID 25158357.
- Dunn FA, Wong RO (2014). "Wiring patterns in the mouse retina: collecting evidence across the connectome, physiology and light microscopy". J Physiol. 592 (22): 4809–4823. doi:10.1113/jphysiol.2014.277228. PMC 4259528. PMID 25172948.
- Audo I, Robson AG, Holder GE, Moore AT (2008). "The negative ERG: clinical phenotypes and disease mechanisms of inner retinal dysfunction". Surv Ophthalmol. 53 (1): 16–40. doi:10.1016/j.survophthal.2007.10.010. PMID 18191655.
- Bech-Hansen N, Naylor M, Maybaum T, Sparkes R, Koop B, Birch D, Bergen A, Prinsen C, Polomeno R, Gal A, Drack A, Musarella M, Jacobson S, Young R, Weleber R (2000). "Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness". Nat Genet. 26 (3): 319–323. doi:10.1038/81619. PMID 11062471.
- Pusch C, Zeitz C, Brandau O, Pesch K, Achatz H, Feil S, Scharfe C, Maurer J, Jacobi F, Pinckers A, Andreasson S, Hardcastle A, Wissinger B, Berger W, Meindl A (2000). "The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein". Nat Genet. 26 (3): 324–327. doi:10.1038/81627. PMID 11062472.
- Gregg R, Mukhopadhyay S, Candille S, Ball S, Pardue M, McCall M, Peachey N (2003). "Identification of the gene and the mutation responsible for the mouse nob phenotype". Invest Ophthalmol Vis Sci. 44 (1): 378–384. doi:10.1167/iovs.02-0501. PMID 12506099.
- Zito I, Allen L, Patel R, Meindl A, Bradshaw K, Yates J, Bird A, Erskine L, Cheetham M, Webster A, Poopalasundaram S, Moore A, Trump D, Hardcastle A (2003). "Mutations in the CACNA1F and NYX genes in British CSNBX families". Hum Mutat. 21 (2): 169. doi:10.1002/humu.9106. PMID 12552565.
- Zeitz C, Minotti R, Feil S, Mátyás G, Cremers F, Hoyng C, Berger W (2005). "Novel mutations in CACNA1F and NYX in Dutch families with X-linked congenital stationary night blindness". Mol Vis. 11: 179–83. PMID 15761389.
- Xiao X, Jia X, Guo X, Li S, Yang Z, Zhang Q (2006). "CSNB1 in Chinese families associated with novel mutations in NYX". J Hum Genet. 51 (7): 634–640. doi:10.1007/s10038-006-0406-5. PMID 16670814.
- Strom T, Nyakatura G, Apfelstedt-Sylla E, Hellebrand H, Lorenz B, Weber B, Wutz K, Gutwillinger N, Rüther K, Drescher B, Sauer C, Zrenner E, Meitinger T, Rosenthal A, Meindl A (1998). "An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness". Nat Genet. 19 (3): 260–263. doi:10.1038/940. PMID 9662399.
- Bech-Hansen N, Naylor M, Maybaum T, Pearce W, Koop B, Fishman G, Mets M, Musarella M, Boycott K (1998). "Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness". Nat Genet. 19 (3): 264–267. doi:10.1038/947. PMID 9662400.
- McRory J, Hamid J, Doering C, Garcia E, Parker R, Hamming K, Chen L, Hildebrand M, Beedle A, Feldcamp L, Zamponi G, Snutch T (2004). "The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution". J Neurosci. 24 (7): 1707–1718. doi:10.1523/JNEUROSCI.4846-03.2004. PMID 14973233.
- Mansergh F, Orton N, Vessey J, Lalonde M, Stell W, Tremblay F, Barnes S, Rancourt D, Bech-Hansen N (2005). "Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina". Hum Mol Genet. 14 (20): 3035–3046. doi:10.1093/hmg/ddi336. PMID 16155113.
- Boycott K, Maybaum T, Naylor M, Weleber R, Robitaille J, Miyake Y, Bergen A, Pierpont M, Pearce W, Bech-Hansen N (2001). "A summary of 20 CACNA1F mutations identified in 36 families with incomplete X-linked congenital stationary night blindness, and characterization of splice variants". Hum Genet. 108 (2): 91–97. doi:10.1007/s004390100461. PMID 11281458.
- Wutz K, Sauer C, Zrenner E, Lorenz B, Alitalo T, Broghammer M, Hergersberg M, de la Chapelle A, Weber B, Wissinger B, Meindl A, Pusch C (2002). "Thirty distinct CACNA1F mutations in 33 families with incomplete type of XLCSNB and Cacna1f expression profiling in mouse retina". Eur J Hum Genet. 10 (8): 449–456. doi:10.1038/sj.ejhg.5200828. PMID 12111638.
- Nakamura M, Ito S, Terasaki H, Miyake Y (2001). "Novel CACNA1F mutations in Japanese patients with incomplete congenital stationary night blindness". Invest Ophthalmol Vis Sci. 42 (7): 1610–6. PMID 11381068.
- Nakamura M, Ito S, Piao C, Terasaki H, Miyake Y (2003). "Retinal and optic disc atrophy associated with a CACNA1F mutation in a Japanese family". Arch Ophthalmol. 121 (7): 1028–1033. doi:10.1001/archopht.121.7.1028. PMID 12860808.
- Allen L, Zito I, Bradshaw K, Patel R, Bird A, Fitzke F, Yates J, Trump D, Hardcastle A, Moore A (2003). "Genotype-phenotype correlation in British families with X linked congenital stationary night blindness". Br J Ophthalmol. 87 (11): 1413–1420. doi:10.1136/bjo.87.11.1413. PMC 1771890. PMID 14609846.
- Hoda J, Zaghetto F, Koschak A, Striessnig J (2005). "Congenital stationary night blindness type 2 mutations S229P, G369D, L1068P, and W1440X alter channel gating or functional expression of Ca(v)1.4 L-type Ca2+ channels". J Neurosci. 25 (1): 252–259. doi:10.1523/JNEUROSCI.3054-04.2005. PMID 15634789.
- Hoda J, Zaghetto F, Singh A, Koschak A, Striessnig J (2006). "Effects of congenital stationary night blindness type 2 mutations R508Q and L1364H on Cav1.4 L-type Ca2+ channel function and expression". J Neurochem. 96 (6): 1648–1658. doi:10.1111/j.1471-4159.2006.03678.x. PMID 16476079.
- Hemara-Wahanui A, Berjukow S, Hope C, Dearden P, Wu S, Wilson-Wheeler J, Sharp D, Lundon-Treweek P, Clover G, Hoda J, Striessnig J, Marksteiner R, Hering S, Maw M (2005). "A CACNA1F mutation identified in an X-linked retinal disorder shifts the voltage dependence of Cav1.4 channel activation". Proc Natl Acad Sci USA. 102 (21): 7553–7558. doi:10.1073/pnas.0501907102. PMC 1140436. PMID 15897456.
- Jacobi F, Hamel C, Arnaud B, Blin N, Broghammer M, Jacobi P, Apfelstedt-Sylla E, Pusch C (2003). "A novel CACNA1F mutation in a french family with the incomplete type of X-linked congenital stationary night blindness". Am J Ophthalmol. 135 (5): 733–736. doi:10.1016/S0002-9394(02)02109-8. PMID 12719097.
- Pere Garriga, and Joan Manyosa. The eye photoreceptor protein rhodopsin. Structural implications for retinal disease. Volume 528, Issues 1–3, 25 September 2002, Pages 17–22.
- V.R. Rao, G.B. Cohen and D.D. Oprian Nature 367 (1994), pp. 639–642.
- N. al-Jandal, G.J. Farrar, A.S. Kiang, M.M. Humphries, N. Bannon, J.B. Findlay, P. Humphries and P.F. Kenna Hum. Mutat. 13 (1999), pp. 75–81.
- T.P. Dryja, E.L. Berson, V.R. Rao and D.D. Oprian Nat. Genet. 4 (1993), pp. 280–283.
- P.A. Sieving, J.E. Richards, F. Naarendorp, E.L. Bingham, K. Scott and M. Alpern Proc. Natl. Acad. Sci. USA 92 (1995), pp. 880–884.
External links
| Classification | |
|---|---|
| External resources |