ABCA4
ATP-binding cassette, sub-family A (ABC1), member 4, also known as ABCA4 or ABCR, is a protein which in humans is encoded by the ABCA4 gene.[5][6][7]


ABCA4 is a member of the ATP-binding cassette transporter gene sub-family A (ABC1) found exclusively in multicellular eukaryotes.[5] The gene was first cloned and characterized in 1997 as a gene that causes Stargardt disease, an autosomal recessive disease that causes macular degeneration.[8] The ABCA4 gene transcribes a large retina-specific protein with two transmembrane domains (TMD), two glycosylated extracellular domains (ECD), and two nucleotide-binding domains (NBD). The ABCA4 protein is almost exclusively expressed in retina localizing in outer segment disk edges of rod photoreceptors.[9]
Structure
Previously known as the photoreceptor rim protein RmP or ABCR, the recently proposed ABCA4 structure consists of two transmembrane domains (TMDs), two large glycosylated extracytosolic domains (ECD), and two internal nucleotide binding domains (NBDs). One TMD spans across membranes with six units of protein linked together to form a domain. The TMDs are usually not conserved across genomes due to its specificity and diversity in function as channels or ligand-binding controllers. However, NBDs are highly conserved across different genomes—an observation consistent with which it binds and hydrolyzes ATP. NBD binds adenosine triphosphate molecules (ATP) to utilize the high-energy inorganic phosphate to carry out change in conformation of the ABC transporter. Transcribed ABCA4 forms into a heterodimer: the two dimerized compartments of the channel are different from each other. When TMDs are situated in a membrane, they form a barrel-like structure permeable to retinoid ligands and control channel access to its binding sites.[10] Once an ATP is hydrolized at the NBDs of the channel, NBDs are brought together to tilt and modify TMDs to modulate ligand binding to the channel.[11] A recently proposed model of retinoid transfer occurring as a result of alternating exposure of external and internal TMD ligand binding sites, all controlled by binding of ATP, is based on recent structural analyses of bacterial ABC transporters.
Function
ABCR is localized to outer segment disk edges of rods and cones. ABCR is expressed much less than rhodopsin, approximately at 1:120. Comparisons between mammalian ABCA4 to other ABCs, cellular localization of ABCA4, and analyses of ABCA4 knockout mice suggest that ABCA4 may function as an inward-directed retinoid flippase.[12] Flippase is a transmembrane protein that "flips" its conformation to transport materials across a membrane. In the case of ABCA4, the flippase facilitates transfer of N-retinyl-phosphatidylethanolamine (NR-PE), a covalent adduct of all-trans retinaldehyde (ATR) with phosphatidylethanolamine (PE), trapped inside the disk as charged species out to the cytoplasmic surface.[13] Once transported, ATR is reduced to vitamin A and then transferred to retinal pigment epithelium to be recycled into 11-cis-retinal. This alternating access-release model for ABCA4 has four steps: (1) binding of ATP to an NBD to bring two NBDs together and expose outer vestibule high affinity binding site located in TMD, (2) binding of NR-PE/ATR on extracellular side of the channel, (3) ATP hydrolysis promoting gate opening and movement of NR-PE/ATR across the membrane to the low-affinity binding site on the intracellular portion of TMD, and (4) release of adenosine diphosphate (ADP) and inorganic phosphate (Pi) to release the bound ligand. The channel is then ready to transfer another molecule of NR-PE/ATR again.
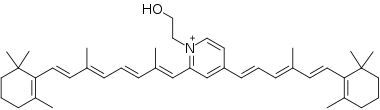
The ABCR -/- knockout mouse has delayed dark adaptation but normal final rod threshold relative to controls.[12] This suggests bulk transmembrane diffusion pathways that remove ATR/NR-PE from extracellular membranes. After bleaching the retina with strong light, ATR/NR-PE accumulates significantly in outer segments. This accumulation leads to formation of toxic cationic bis-pyridinium salt, N-retinylidene-N-retinyl-ethanolamine (A2E), which causes human dry and wet age-related macular degeneration.[14] From this experiment, it was concluded that ABCR has a significant role in clearing accumulation of ATR/NR-PE to prevent formation of A2E in extracellular photoreceptor surfaces during bleach recovery.
Clinical significance
Mutations in ABCA4 gene are known to cause the autosomal-recessive disease Stargardt macular dystrophy (STGD), which is a hereditary juvenile macular degeneration disease causing progressive loss of photoreceptor cells. STGD is characterized by reduced visual acuity and color vision, loss of central (macular) vision, delayed dark adaptation, and accumulation of autoflourescent RPE lipofuscin.[14] Removal of NR-PE/ATR appears to be significant in normal bleach recovery and to mitigate persistent opsin signaling that causes photoreceptors to degenerate. ABCA4 also mitigates long-term effects of accumulation of ATR that results in irreversible ATR binding to a second molecule of ATR and NR-PE to form dihydro-N-retinylidene-N-retinyl-phosphatidyl-ethanolamine (A2PE-H2). A2PE-H2 traps ATR and accumulates in outer segments to further oxidize into N-retinylidene-N-retinyl-phosphatidyl-ethanolamine (A2PE). After diurnal disk-shedding and phagocytosis of outer segment by RPE cells, A2PE is hydrolyzed inside the RPE phagolysosome to form A2E.[14] Accumulation of A2E causes toxicity at the primary RPE level and secondary photoreceptor destruction in macular degenerations.
Additional diseases that may link to mutations in ABCA4 include fundus flavimaculatus, cone-rod dystrophy, retinitis pigmentosa, and age-related macular degeneration.
The GENEVA Cleft Consortium study first identified ABCA4 as being associated with cleft lip and/or cleft palate with multiple markers giving evidence of linkage and association at the genome-wide significance level.[15] Although SNPs in this gene are associated with cleft lip/palate there is no functional or expression data to support it as the causal gene which may, instead, lie in a region adjacent to ABCA4.[16] A combination of genome wide association, rare coding sequence variants, craniofacial specific expression, and interactions with IRF6 support a role for the adjacent ARHGAP29 gene to be the likely causal gene playing a role in nonsyndromic cleft lip and/or palate.[17]
See also
References
- GRCh38: Ensembl release 89: ENSG00000198691 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000028125 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Entrez Gene: ABCA4 ATP-binding cassette, sub-family A (ABC1), member 4".
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M, Lupski JR (March 1997). "A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy". Nature Genetics. 15 (3): 236–46. doi:10.1038/ng0397-236. PMID 9054934.
- Nasonkin I, Illing M, Koehler MR, Schmid M, Molday RS, Weber BH (January 1998). "Mapping of the rod photoreceptor ABC transporter (ABCR) to 1p21-p22.1 and identification of novel mutations in Stargardt's disease". Human Genetics. 102 (1): 21–6. doi:10.1007/s004390050649. PMID 9490294.
- Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, Peiffer A, Zabriskie NA, Li Y, Hutchinson A, Dean M, Lupski JR, Leppert M (September 1997). "Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration". Science. 277 (5333): 1805–7. doi:10.1126/science.277.5333.1805. PMID 9295268.
- Sun H, Nathans J (2000). "ABCR: rod photoreceptor-specific ABC transporter responsible for Stargardt disease". Methods in Enzymology. 315: 879–97. doi:10.1016/S0076-6879(00)15888-4. ISBN 978-0-12-182216-3. PMID 10736747.
- van Meer G, Halter D, Sprong H, Somerharju P, Egmond MR (February 2006). "ABC lipid transporters: extruders, flippases, or flopless activators?". FEBS Letters. 580 (4): 1171–7. doi:10.1016/j.febslet.2005.12.019. hdl:1874/19996. PMID 16376334.
- Sullivan JM (November 2009). "Focus on molecules: ABCA4 (ABCR)--an import-directed photoreceptor retinoid flipase". Experimental Eye Research. 89 (5): 602–3. doi:10.1016/j.exer.2009.03.005. PMC 3371273. PMID 19306869.
- Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH (July 1999). "Insights into the function of Rim protein in photoreceptors and etiology of Stargardt's disease from the phenotype in abcr knockout mice". Cell. 98 (1): 13–23. doi:10.1016/S0092-8674(00)80602-9. PMID 10412977.
- Molday RS, Beharry S, Ahn J, Zhong M (2006). "Binding of N-retinylidene-PE to ABCA4 and a model for its transport across membranes". Advances in Experimental Medicine and Biology. 572: 465–70. doi:10.1007/0-387-32442-9_64. ISBN 978-0-387-28464-4. PMID 17249610.
- Maeda A, Maeda T, Golczak M, Palczewski K (September 2008). "Retinopathy in mice induced by disrupted all-trans-retinal clearance". The Journal of Biological Chemistry. 283 (39): 26684–93. doi:10.1074/jbc.M804505200. PMC 2546559. PMID 18658157.
- Dixon MJ, Marazita ML, Beaty TH, Murray JC (March 2011). "Cleft lip and palate: understanding genetic and environmental influences". Nature Reviews Genetics. 12 (3): 167–78. doi:10.1038/nrg2933. PMC 3086810. PMID 21331089.
- Beaty TH, Ruczinski I, Murray JC, Marazita ML, Munger RG, Hetmanski JB, Murray T, Redett RJ, Fallin MD, Liang KY, Wu T, Patel PJ, Jin SC, Zhang TX, Schwender H, Wu-Chou YH, Chen PK, Chong SS, Cheah F, Yeow V, Ye X, Wang H, Huang S, Jabs EW, Shi B, Wilcox AJ, Lie RT, Jee SH, Christensen K, Doheny KF, Pugh EW, Ling H, Scott AF (September 2011). "Evidence for gene-environment interaction in a genome wide study of nonsyndromic cleft palate". Genetic Epidemiology. 35 (6): 469–78. doi:10.1002/gepi.20595. PMC 3180858. PMID 21618603.
- Leslie EJ, Mansilla MA, Biggs LC, Schuette K, Bullard S, Cooper M, Dunnwald M, Lidral AC, Marazita ML, Beaty TH, Murray JC (November 2012). "Expression and mutation analyses implicate ARHGAP29 as the etiologic gene for the cleft lip with or without cleft palate locus identified by genome-wide association on chromosome 1p22". Birth Defects Research. Part A, Clinical and Molecular Teratology. 94 (11): 934–42. doi:10.1002/bdra.23076. PMC 3501616. PMID 23008150.
Further reading
- MacDonald IM (June 2005). "Genetic aspects of age-related macular degeneration". Canadian Journal of Ophthalmology. 40 (3): 288–92. doi:10.1016/S0008-4182(05)80071-7. PMID 15947798.
- Bonaldo MF, Lennon G, Soares MB (September 1996). "Normalization and subtraction: two approaches to facilitate gene discovery". Genome Research. 6 (9): 791–806. doi:10.1101/gr.6.9.791. PMID 8889548.
- Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M, Lupski JR (March 1997). "A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy". Nature Genetics. 15 (3): 236–46. doi:10.1038/ng0397-236. PMID 9054934.
- Martínez-Mir A, Bayés M, Vilageliu L, Grinberg D, Ayuso C, del Río T, García-Sandoval B, Bussaglia E, Baiget M, Gonzàlez-Duarte R, Balcells S (February 1997). "A new locus for autosomal recessive retinitis pigmentosa (RP19) maps to 1p13-1p21". Genomics. 40 (1): 142–6. doi:10.1006/geno.1996.4528. hdl:10261/39369. PMID 9070931.
- Azarian SM, Travis GH (June 1997). "The photoreceptor rim protein is an ABC transporter encoded by the gene for recessive Stargardt's disease (ABCR)". FEBS Letters. 409 (2): 247–52. doi:10.1016/S0014-5793(97)00517-6. PMID 9202155.
- Sun H, Nathans J (September 1997). "Stargardt's ABCR is localized to the disc membrane of retinal rod outer segments". Nature Genetics. 17 (1): 15–6. doi:10.1038/ng0997-15. PMID 9288089.
- Allikmets R (September 1997). "A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy". Nature Genetics. 17 (1): 122. doi:10.1038/ng0997-122a. PMID 9288113.
- Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, Peiffer A, Zabriskie NA, Li Y, Hutchinson A, Dean M, Lupski JR, Leppert M (September 1997). "Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration". Science. 277 (5333): 1805–7. doi:10.1126/science.277.5333.1805. PMID 9295268.
- Martínez-Mir A, Paloma E, Allikmets R, Ayuso C, del Rio T, Dean M, Vilageliu L, Gonzàlez-Duarte R, Balcells S (January 1998). "Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR". Nature Genetics. 18 (1): 11–2. doi:10.1038/ng0198-11. hdl:10261/39477. PMID 9425888.
- Cremers FP, van de Pol DJ, van Driel M, den Hollander AI, van Haren FJ, Knoers NV, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Deutman AF, Hoyng CB (March 1998). "Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR". Human Molecular Genetics. 7 (3): 355–62. doi:10.1093/hmg/7.3.355. PMID 9466990.
- Nasonkin I, Illing M, Koehler MR, Schmid M, Molday RS, Weber BH (January 1998). "Mapping of the rod photoreceptor ABC transporter (ABCR) to 1p21-p22.1 and identification of novel mutations in Stargardt's disease". Human Genetics. 102 (1): 21–6. doi:10.1007/s004390050649. PMID 9490294.
- Gerber S, Rozet JM, van de Pol TJ, Hoyng CB, Munnich A, Blankenagel A, Kaplan J, Cremers FP (February 1998). "Complete exon-intron structure of the retina-specific ATP binding transporter gene (ABCR) allows the identification of novel mutations underlying Stargardt disease". Genomics. 48 (1): 139–42. doi:10.1006/geno.1997.5164. PMID 9503029.
- Azarian SM, Megarity CF, Weng J, Horvath DH, Travis GH (June 1998). "The human photoreceptor rim protein gene (ABCR): genomic structure and primer set information for mutation analysis". Human Genetics. 102 (6): 699–705. doi:10.1007/s004390050765. PMID 9703434.
- Rozet JM, Gerber S, Souied E, Perrault I, Châtelin S, Ghazi I, Leowski C, Dufier JL, Munnich A, Kaplan J (1998). "Spectrum of ABCR gene mutations in autosomal recessive macular dystrophies". European Journal of Human Genetics. 6 (3): 291–5. doi:10.1038/sj.ejhg.5200221. PMID 9781034.
- Lewis RA, Shroyer NF, Singh N, Allikmets R, Hutchinson A, Li Y, Lupski JR, Leppert M, Dean M (February 1999). "Genotype/Phenotype analysis of a photoreceptor-specific ATP-binding cassette transporter gene, ABCR, in Stargardt disease". American Journal of Human Genetics. 64 (2): 422–34. doi:10.1086/302251. PMC 1377752. PMID 9973280.
- Sun H, Molday RS, Nathans J (March 1999). "Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease". The Journal of Biological Chemistry. 274 (12): 8269–81. doi:10.1074/jbc.274.12.8269. PMID 10075733.
- Maugeri A, van Driel MA, van de Pol DJ, Klevering BJ, van Haren FJ, Tijmes N, Bergen AA, Rohrschneider K, Blankenagel A, Pinckers AJ, Dahl N, Brunner HG, Deutman AF, Hoyng CB, Cremers FP (April 1999). "The 2588G-->C mutation in the ABCR gene is a mild frequent founder mutation in the Western European population and allows the classification of ABCR mutations in patients with Stargardt disease". American Journal of Human Genetics. 64 (4): 1024–35. doi:10.1086/302323. PMC 1377826. PMID 10090887.
- Fishman GA, Stone EM, Grover S, Derlacki DJ, Haines HL, Hockey RR (April 1999). "Variation of clinical expression in patients with Stargardt dystrophy and sequence variations in the ABCR gene". Archives of Ophthalmology. 117 (4): 504–10. doi:10.1001/archopht.117.4.504. PMID 10206579.
- Körschen HG, Beyermann M, Müller F, Heck M, Vantler M, Koch KW, Kellner R, Wolfrum U, Bode C, Hofmann KP, Kaupp UB (August 1999). "Interaction of glutamic-acid-rich proteins with the cGMP signalling pathway in rod photoreceptors". Nature. 400 (6746): 761–6. doi:10.1038/23468. PMID 10466724.
- Zhang K, Garibaldi DC, Kniazeva M, Albini T, Chiang MF, Kerrigan M, Sunness JS, Han M, Allikmets R (December 1999). "A novel mutation in the ABCR gene in four patients with autosomal recessive Stargardt disease". American Journal of Ophthalmology. 128 (6): 720–4. doi:10.1016/S0002-9394(99)00236-6. PMID 10612508.
External links
- GeneReviews/NIH/NCBI/UW entry on Retinitis Pigmentosa Overview
- ABCA4+protein,+human at the US National Library of Medicine Medical Subject Headings (MeSH)
- ABCA4 human gene location in the UCSC Genome Browser.
- ABCA4 human gene details in the UCSC Genome Browser.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.