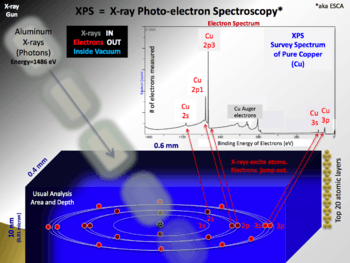
X-ray photoelectron spectroscopy
X-ray photoelectron spectroscopy (XPS) is a surface-sensitive quantitative spectroscopic technique based on the photoelectric effect that can identify the elements that exist within a material (elemental composition) or are covering its surface, as well as their chemical state, and the overall electronic structure and density of the electronic states in the material. XPS is a powerful measurement technique because it not only shows what elements are present, but also what other elements they are bonded to. The technique can be used in line profiling of the elemental composition across the surface, or in depth profiling when paired with ion-beam etching. It is often applied to study chemical processes in the materials in their as-received state or after cleavage, scraping, exposure to heat, reactive gasses or solutions, ultraviolet light, or during ion implantation.
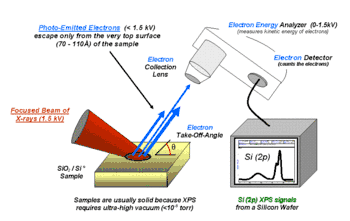
XPS belongs to the family of photoemission spectroscopies in which electron population spectra are obtained by irradiating a material with a beam of X-rays. Material properties are inferred from the measurement of the kinetic energy and the number of the ejected electrons. XPS requires high vacuum (residual gas pressure p ~ 10−6 Pa) or ultra-high vacuum (p < 10−7 Pa) conditions, although a current area of development is ambient-pressure XPS, in which samples are analyzed at pressures of a few tens of millibar.
When laboratory X-ray sources are used, XPS easily detects all elements except hydrogen and helium. Detection limit is in the parts per thousand range, but parts per million (ppm) are achievable with long collection times and concentration at top surface.
XPS is routinely used to analyze inorganic compounds, metal alloys,[1] semiconductors,[2] polymers, elements, catalysts,[3][4][5][6] glasses, ceramics, paints, papers, inks, woods, plant parts, make-up, teeth, bones, medical implants, bio-materials,[7] coatings,[8] viscous oils, glues, ion-modified materials and many others. Somewhat less routinely XPS is used to analyze the hydrated forms of materials such as hydrogels and biological samples by freezing the them in their hydrated state in an ultrapure environment, and allowing multilayers of ice to sublime away prior to analysis.
Basic physics
Because the energy of an X-ray with particular wavelength is known (for Al Kα X-rays, Ephoton = 1486.7 eV), and because the emitted electrons' kinetic energies are measured, the electron binding energy of each of the emitted electrons can be determined by using the photoelectric effect equation:

where Ebinding is the binding energy (BE) of the electron measured relative to the chemical potential, Ephoton is the energy of the X-ray photons being used, Ekinetic is the kinetic energy of the electron as measured by the instrument and is the work function for the specific surface of the material, which in real measurements includes a small correction by the instrument's work function because of the contact potential. This equation is essentially a conservation of energy equation. The work function-like term can be thought of as an adjustable instrumental correction factor that accounts for the few eV of kinetic energy given up by the photoelectron as it gets emitted from the bulk and absorbed by the detector. It is a constant that rarely needs to be adjusted in practice.

History

In 1887, Heinrich Rudolf Hertz discovered but could not explain the photoelectric effect, which was later explained in 1905 by Albert Einstein (Nobel Prize in Physics 1921). Two years after Einstein's publication, in 1907, P.D. Innes experimented with a Röntgen tube, Helmholtz coils, a magnetic field hemisphere (an electron kinetic energy analyzer), and photographic plates, to record broad bands of emitted electrons as a function of velocity, in effect recording the first XPS spectrum. Other researchers, including Henry Moseley, Rawlinson and Robinson, independently performed various experiments to sort out the details in the broad bands. After WWII, Kai Siegbahn and his research group in Uppsala (Sweden) developed several significant improvements in the equipment, and in 1954 recorded the first high-energy-resolution XPS spectrum of cleaved sodium chloride (NaCl), revealing the potential of XPS.[9] A few years later in 1967, Siegbahn published a comprehensive study of XPS, bringing instant recognition of the utility of XPS, which he referred to as Electron Spectroscopy for Chemical Analysis (ESCA). In cooperation with Siegbahn, a small group of engineers (Mike Kelly, Charles Bryson, Lavier Faye, Robert Chaney) at Hewlett-Packard in the USA, produced the first commercial monochromatic XPS instrument in 1969. Siegbahn received the Nobel Prize for Physics in 1981, to acknowledge his extensive efforts to develop XPS into a useful analytical tool.[10] In parallel with Siegbahn's work, David Turner at Imperial College London (and later at Oxford University) developed ultraviolet photoelectron spectroscopy (UPS) for molecular species using helium lamps.[11]
Measurement
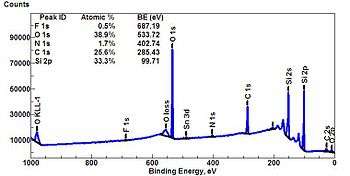
A typical XPS spectrum is a plot of the number of electrons detected at a specific binding energy. Each element produces a set of characteristic XPS peaks. These peaks correspond to the electron configuration of the electrons within the atoms, e.g., 1s, 2s, 2p, 3s, etc. The number of detected electrons in each peak is directly related to the amount of element within the XPS sampling volume. To generate atomic percentage values, each raw XPS signal is corrected by dividing the intensity by a relative sensitivity factor (RSF), and normalized over all of the elements detected. Since hydrogen is not detected, these atomic percentages exclude hydrogen.
Quantitative accuracy and precision
XPS is widely used to generate an empirical formula because it readily yields excellent quantitative accuracy from homogeneous solid-state materials. Absolute quantification requires the use of certified (or independently verified) standard samples, and is generally more challenging, and less common. Relative quantification involves comparisons between several samples in a set for which one or more analytes are varied while all other components (the sample matrix) are held constant. Quantitative accuracy depends on several parameters such as: signal-to-noise ratio, peak intensity, accuracy of relative sensitivity factors, correction for electron transmission function, surface volume homogeneity, correction for energy dependence of electron mean free path, and degree of sample degradation due to analysis. Under optimal conditions, the quantitative accuracy of the atomic percent (at%) values calculated from the major XPS peaks is 90-95% for each peak. The quantitative accuracy for the weaker XPS signals, that have peak intensities 10-20% of the strongest signal, are 60-80% of the true value, and depend upon the amount of effort used to improve the signal-to-noise ratio (for example by signal averaging). Quantitative precision (the ability to repeat a measurement and obtain the same result) is an essential consideration for proper reporting of quantitative results. Standard statistical tests, such as the Student's t test for comparison of means, should be used to determine confidence levels in the average value from a set of replicate measurements, and when comparing the average values of two or more different sets of results. In general, a p value (an output of the Student's t test) of 0.05 or less indicates a level of confidence (95%) that is accepted in the field as significant.
Detection limits
Detection limits may vary greatly with the cross section of the core state of interest and the background signal level. In general, photoelectron cross sections increase with atomic number. The background increases with the atomic number of the matrix constituents as well as the binding energy, because of secondary emitted electrons. For example in the case of gold on silicon where the high cross section Au4f peak is at a higher kinetic energy than the major silicon peaks, it sits on a very low background and detection limits of 1ppm or better may be achieved with reasonable acquisition times. Conversely for silicon on gold, where the modest cross section Si2p line sits on the large background below the Au4f lines, detection limits would be much worse for the same acquisition time. Detection limits are often quoted as 0.1–1.0 % atomic percent (0.1% = 1 part per thousand = 1000 ppm) for practical analyses, but lower limits may be achieved in many circumstances.
Degradation during analysis
Degradation depends on the sensitivity of the material to the wavelength of X-rays used, the total dose of the X-rays, the temperature of the surface and the level of the vacuum. Metals, alloys, ceramics and most glasses are not measurably degraded by either non-monochromatic or monochromatic X-rays. Some, but not all, polymers, catalysts, certain highly oxygenated compounds, various inorganic compounds and fine organics are. Non-monochromatic X-ray sources produce a significant amount of high energy Bremsstrahlung X-rays (1–15 keV of energy) which directly degrade the surface chemistry of various materials. Non-monochromatic X-ray sources also produce a significant amount of heat (100 to 200 °C) on the surface of the sample because the anode that produces the X-rays is typically only 1 to 5 cm (2 in) away from the sample. This level of heat, when combined with the Bremsstrahlung X-rays, acts to increase the amount and rate of degradation for certain materials. Monochromatised X-ray sources, because they are farther away (50–100 cm) from the sample, do not produce noticeable heat effects. In those, a quartz monochromator system diffracts the Bremsstrahlung X-rays out of the X-ray beam, which means the sample is only exposed to one narrow band of X-ray energy. For example, if aluminum K-alpha X-rays are used, the intrinsic energy band has a FWHM of 0.43 eV, centered on 1,486.7 eV (E/ΔE = 3,457). If magnesium K-alpha X-rays are used, the intrinsic energy band has a FWHM of 0.36 eV, centered on 1,253.7 eV (E/ΔE = 3,483). These are the intrinsic X-ray line widths; the range of energies to which the sample is exposed depends on the quality and optimization of the X-ray monochromator. Because the vacuum removes various gases (e.g., O2, CO) and liquids (e.g., water, alcohol, solvents, etc.) that were initially trapped within or on the surface of the sample, the chemistry and morphology of the surface will continue to change until the surface achieves a steady state. This type of degradation is sometimes difficult to detect.
Measured area
Measured area depends on instrument design. The minimum analysis area ranges from 10 to 200 micrometres. Largest size for a monochromatic beam of X-rays is 1–5 mm. Non-monochromatic beams are 10–50 mm in diameter. Spectroscopic image resolution levels of 200 nm or below has been achieved on latest imaging XPS instruments using synchrotron radiation as X-ray source.
Sample size limits
Instruments accept small (mm range) and large samples (cm range), e.g. wafers. Limiting factor is the design of the sample holder, the sample transfer, and the size of the vacuum chamber. Large samples are laterally moved in x and y direction to analyse a larger area.
Analysis time
Typically ranging 1–20 minutes for a broad survey scan that measures the amount of all detectable elements, typically 1–15 minutes for high resolution scan that reveal chemical state differences (for a high signal/noise ratio for count area result often requires multiple sweeps of the region of interest), 1–4 hours for a depth profile that measures 4–5 elements as a function of etched depth (this process time can vary the most as many factors will play a role).
Surface sensitivity
XPS detects only electrons that have actually escaped from the sample into the vacuum of the instrument. In order to escape from the sample, a photoelectron must travel through the sample. Photo-emitted electrons can undergo inelastic collisions, recombination, excitation of the sample, recapture or trapping in various excited states within the material, all of which can reduce the number of escaping photoelectrons. These effects appear as an exponential attenuation function as the depth increases, making the signals detected from analytes at the surface much stronger than the signals detected from analytes deeper below the sample surface. Thus, the signal measured by XPS is an exponentially surface-weighted signal, and this fact can be used to estimate analyte depths in layered materials.
Chemical states and chemical shift
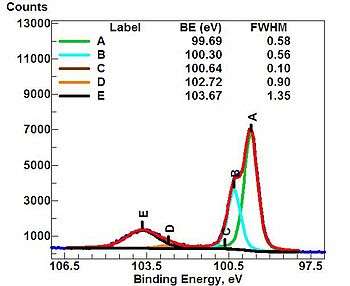
The ability to produce chemical state information, i.e. the local bonding environment of an atomic species in question from the topmost few nanometers of the sample makes XPS a unique and valuable tool for understanding the chemistry of the surface. The local bonding environment is affected by the formal oxidation state, the identity of its nearest-neighbor atoms, and its bonding hybridization to the nearest-neighbor or next-nearest-neighbor atoms. For example, while the nominal binding energy of the C1s electron is 284.6 eV, subtle but reproducible shifts in the actual binding energy, the so-called chemical shift (analogous to NMR spectroscopy), provide the chemical state information.
Chemical-state analysis is widely used for carbon. It reveals the presence or absence of the chemical states of carbon, in approximate order of increasing binding energy, as: carbide (-C2−), silane (-Si-CH3), methylene/methyl/hydrocarbon (-CH2-CH2-, CH3-CH2-, and -CH=CH-), amine (-CH2-NH2), alcohol (-C-OH), ketone (-C=O), organic ester (-COOR), carbonate (-CO32−), monofluoro-hydrocarbon (-CFH-CH2-), difluoro-hydrocarbon (-CF2-CH2-), and trifluorocarbon (-CH2-CF3), to name but a few.
Chemical state analysis of the surface of a silicon wafer reveals chemical shifts due to different formal oxidation states, such as: n-doped silicon and p-doped silicon (metallic silicon), silicon suboxide (Si2O), silicon monoxide (SiO), Si2O3, and silicon dioxide (SiO2). An example of this is seen in the figure "High-resolution spectrum of an oxidized silicon wafer in the energy range of the Si 2p signal".Routine limits
Instrumentation
The main components of an XPS system are the source of X-rays, an ultra-high vacuum (UHV) chamber with mu-metal magnetic shielding, an electron collection lens, an electron energy analyzer, an electron detector system, a sample introduction chamber, sample mounts, a sample stage with the ability to heat or cool the sample, and a set of stage manipulators.
The most prevalent electron spectrometer for XPS is the hemispherical electron analyzer. They have high energy resolution and spatial selection of the emitted electrons. Sometimes, however, much simpler electron energy filters - the cylindrical mirror analyzers are used, most often for checking the elemental composition of the surface. They represent a trade-off between the need for high count rates and high angular/energy resolution. This type consists of two co-axial cylinders placed in front of the sample, the inner one being held at a positive potential, while the outer cylinder is held at a negative potential. Only the electrons with the right energy can pass through this setup and are detected at the end. The count rates are high but the resolution (both in energy and angle) is poor.
Electrons are detected using electron multipliers: a single channeltron for single energy detection, or arrays of channeltrons and microchannel plates for parallel acquisition. These devices consists of a glass channel with a resistive coating on the inside. A high voltage is applied between the front and the end. An incoming electron is accelerated to the wall, where it removes more electrons, in such a way that an electron avalanche is created, until a measurable current pulse is obtained.
Laboratory based XPS
In laboratory systems, either 10-30 mm beam diameter non-monochromatic Al Kα or Mg Kα anode radiation is used, or a focused 20-500 micrometer diameter beam single wavelength Al Kα monochromatised radiation. Monochromatic Al Kα X-rays are normally produced by diffracting and focusing a beam of non-monochromatic X-rays off of a thin disc of natural, crystalline quartz with a <1010> orientation. The resulting wavelength is 8.3386 angstroms (0.83386 nm) which corresponds to a photon energy of 1486.7 eV. Aluminum Kα X-rays have an intrinsic full width at half maximum (FWHM) of 0.43 eV, centered on 1486.7 eV (E/ΔE = 3457). For a well–optimized monochromator, the energy width of the monochromated aluminum Kα X-rays is 0.16 eV, but energy broadening in common electron energy analyzers (spectrometers) produces an ultimate energy resolution on the order of FWHM=0.25 eV which, in effect, is the ultimate energy resolution of most commercial systems. When working under practical, everyday conditions, high energy-resolution settings will produce peak widths (FWHM) between 0.4–0.6 eV for various pure elements and some compounds. For example, in a spectrum obtained in 1 minute at a pass energy of 20 eV using monochromated aluminum Kα X-rays, the Ag 3d5/2 peak for a clean silver film or foil will typically have a FWHM of 0.45 eV. Non-monochromatic magnesium X-rays have a wavelength of 9.89 angstroms (0.989 nm) which corresponds to a photon energy of 1253 eV. The energy width of the non-monochromated X-ray is roughly 0.70 eV, which, in effect is the ultimate energy resolution of a system using non-monochromatic X-rays. Non-monochromatic X-ray sources do not use any crystals to diffract the X-rays which allows all primary X-rays lines and the full range of high-energy Bremsstrahlung X-rays (1–12 keV) to reach the surface. The ultimate energy resolution (FWHM) when using a non-monochromatic Mg Kα source is 0.9–1.0 eV, which includes some contribution from spectrometer-induced broadening.
Synchrotron based XPS
A breakthrough has been brought about in the last decades by the development of large scale synchrotron radiation facilities. Here, bunches of relativistic electrons kept in orbit inside a storage ring are accelerated through bending magnets or insertion devices like wigglers and undulators to produce a high brilliance and high flux photon beam. The beam is orders of magnitude more intense and better collimated than typically produced by anode-based sources. Synchrotron radiation is also tunable over a wide wavelength range, and can be made polarized in several distinct ways. This way, photon can be selected yielding optimum photoionization cross-sections for probing a particular core level. The high photon flux, in addition, makes it possible to perform XPS experiments also from low density atomic species, such as molecular and atomic adsorbates.
Data processing
Peak identification
The number of peaks produced by a single element varies from 1 to more than 20. Tables of binding energies that identify the shell and spin-orbit of each peak produced by a given element are included with modern XPS instruments, and can be found in various handbooks and websites.[12][13] Because these experimentally determined energies are characteristic of specific elements, they can be directly used to identify experimentally measured peaks of a material with unknown elemental composition.
Before beginning the process of peak identification, the analyst must determine if the binding energies of the unprocessed survey spectrum (0-1400 eV) have or have not been shifted due to a positive or negative surface charge. This is most often done by looking for two peaks that are due to the presence of carbon and oxygen.
Charge referencing insulators
Charge referencing is needed when a sample suffers a charge induced shift of experimental binding energies to obtain meaningful binding energies from both wide-scan, high sensitivity (low energy resolution) survey spectra (0-1100 eV), and also narrow-scan, chemical state (high energy resolution) spectra. Charge induced shifting is normally due to a modest excess of low voltage (-1 to -20 eV) electrons attached to the surface, or a modest shortage of electrons (+1 to +15 eV) within the top 1-12 nm of the sample caused by the loss of photo-emitted electrons. If, by chance, the charging of the surface is excessively positive, then the spectrum might appear as a series of rolling hills, not sharp peaks as shown in the example spectrum.
Charge referencing is performed by adding a Charge Correction Factor to each of the experimentally measured peaks. Since various hydrocarbon species appear on all air-exposed surfaces, the binding energy of the hydrocarbon C (1s) XPS peak is used to charge correct all energies obtained from non-conductive samples or conductors that have been deliberately insulated from the sample mount. The peak is normally found between 284.5 eV and 285.5 eV. The 284.8 eV binding energy is routinely used as the reference binding energy for charge referencing insulators, so that the charge correction factor is the difference between 284.8 eV and the experimentally measured C (1s) peak position.
Conductive materials and most native oxides of conductors should never need charge referencing. Conductive materials should never be charge referenced unless the topmost layer of the sample has a thick non-conductive film. The charging effect, if needed, can also be compensated by providing suitable low energy charges to the surface by the use of low-voltage (1-20 eV) electron beam from an electron flood gun, UV lights, low-voltage argon ion beam with low-voltage electron beam (1-10 eV), aperture masks, mesh screen with low-voltage electron beams, etc.
Peak-fitting
The process of peak-fitting high energy resolution XPS spectra is a mixture of scientific knowledge and experience. The process is affected by instrument design, instrument components, experimental settings and sample variables. Before starting any peak-fit effort, the analyst performing the peak-fit needs to know if the topmost 15 nm of the sample is expected to be a homogeneous material or is expected to be a mixture of materials. If the top 15 nm is a homogeneous material with only very minor amounts of adventitious carbon and adsorbed gases, then the analyst can use theoretical peak area ratios to enhance the peak-fitting process. Peak fitting results are affected by overall peak widths (at half maximum, FWHM), possible chemical shifts, peak shapes, instrument design factors and experimental settings, as well as sample properties:
- The full width at half maximum (FWHM) values are useful indicators of chemical state changes and physical influences. Their increase may indicate a change in the number of chemical bonds, a change in the sample condition (x-ray damage) or differential charging of the surface (localised differences in the charge state of the surface). However, the FWHM also depends on the detector, and can also increase due to the sample getting charged. When using high energy resolution experiment settings on an XPS equipped with a monochromatic Al K-alpha X-ray source, the FWHM of the major XPS peaks range from 0.3 eV to 1.7 eV. The following is a simple summary of FWHM from major XPS signals: Main metal peaks (e.g. 1s, 2p3, 3d5, 4f7) from pure metals have FWHMs that range from 0.30 eV to 1.0 eV Main metal peaks (e.g. 1s, 2p3, 3d5, 4f7) from binary metal oxides have FWHMs that range from 0.9 eV to 1.7 eV The O (1s) peak from binary metal oxides have FWHMs that, in general, range from 1.0 eV to 1.4 eV The C (1s) peak from adventitious hydrocarbons have FWHMs that, in general, range from 1.0 eV to 1.4 eV
- Chemical shift values depend on the degree of electron bond polarization between nearest-neighbor atoms. A specific chemical shift is the difference in BE values of one specific chemical state versus the BE of one form of the pure element, or of a particular agreed-upon chemical state of that element. Component peaks derived from peak-fitting a raw chemical state spectrum can be assigned to the presence of different chemical states within the sampling volume of the sample.
- Peak shapes depend on instrument parameters, experimental parameters and sample characteristics.
- Instrument design factors include linewidth and purity of X-rays used (monochromatic Al, non-monochromatic Mg, Synchrotron, Ag, Zr), as well as properties of the electron analyzer.
- Settings of the electron analyzer (e.g. pass energy, step size)
- Sample factors that affect the peak fitting are the number of physical defects within the analysis volume (from ion etching, or laser cleaning), and the very physical form of the sample (single crystal, polished, powder, corroded)
Theoretical aspects
Quantum mechanical treatment
When a photoemission event takes place, the following energy conservation rule holds:

where is the photon energy, is the electron BE (with respect to the vacuum level) prior to ionization, and is the kinetic energy of the photoelectron. If reference is taken with respect to the Fermi level (as it is typically done in photoelectron spectroscopy) must be replaced by the sum of the binding energy (BE) relative to the Fermi level, , and the sample work function, .





From the theoretical point of view, the photoemission process from a solid can be described with a semiclassical approach, where the electromagnetic field is still treated classically, while a quantum-mechanical description is used for matter. The one—particle Hamiltonian for an electron subjected to an electromagnetic field is given by:
- ,

where is the electron wave function, is the vector potential of the electromagnetic field and is the unperturbed potential of the solid. In the Coulomb gauge (), the vector potential commutes with the momentum operator (), so that the expression in brackets in the Hamiltonian simplifies to:






Actually, neglecting the term in the Hamiltonian, we are disregarding possible photocurrent contributions.[14] Such effects are generally negligible in the bulk, but may become important at the surface. The quadratic term in can be instead safely neglected, since its contribution in a typical photoemission experiment is about one order of magnitude smaller than that of the first term .

In first-order perturbation approach, the one-electron Hamiltonian can be split into two terms, an unperturbed Hamiltonian , plus an interaction Hamiltonian , which describes the effects of the electromagnetic field:



In time-dependent perturbation theory, for an harmonic or constant perturbation, the transition rate between the initial state and the final state is expressed by Fermi's Golden Rule:


- ,

where and are the eigenvalues of the unperturbed Hamiltonian in the initial and final state, respectively, and is the photon energy. Fermi's Golden Rule uses the approximation that the perturbation acts on the system for an infinite time. This approximation is valid when the time that the perturbation acts on the system is much larger than the time needed for the transition. It should be understood that this equation needs to be integrated with the density of states which gives:[15]




In a real photoemission experiment the ground state core electron BE cannot be directly probed, because the measured BE incorporates both initial state and final state effects, and the spectral linewidth is broadened owing to the finite core-hole lifetime ().

Assuming an exponential decay probability for the core hole in the time domain (), the spectral function will have a Lorentzian shape, with a FWHM (Full Width at Half Maximum) given by:



From the theory of Fourier transforms, and are linked by the indeterminacy relation:

The photoemission event leaves the atom in a highly excited core ionized state, from which it can decay radiatively (fluorescence) or non-radiatively (typically by Auger decay). Besides Lorentzian broadening, photoemission spectra are also affected by a Gaussian broadening, whose contribution can be expressed by

Three main factors enter the Gaussian broadening of the spectra: the experimental energy resolution, vibrational and inhomogeneous broadening. The first effect is caused by the non perfect monochromaticity of the photon beam -which results in a finite bandwidth- and by the limited resolving power of the analyzer. The vibrational component is produced by the excitation of low energy vibrational modes both in the initial and in the final state. Finally, inhomogeneous broadening can originate from the presence of unresolved core level components in the spectrum.
Theory of core level photoemission of electrons
Inelastic mean free path
In a solid, inelastic scattering events also contribute to the photoemission process, generating electron-hole pairs which show up as an inelastic tail on the high BE side of the main photoemission peak. In fact this allows the calculation of electron inelastic mean free path (IMFP). This can be modeled based on the Beer–Lambert law, which states

where is the IMFP and is the axis perpendicular to the sample. In fact it is generally the case that the IMFP is only weakly material dependent, but rather strongly dependent on the photoelectron kinetic energy. Quantitatively we can relate to IMFP by[16][17]




where is the mean atomic diameter as calculated by the density so . The above formula was developed by Seah and Dench.


Plasmonic effects
In some cases, energy loss features due to plasmon excitations are also observed. This can either be a final state effect caused by core hole decay, which generates quantized electron wave excitations in the solid (intrinsic plasmons), or it can be due to excitations induced by photoelectrons travelling from the emitter to the surface (extrinsic plasmons). Due to the reduced coordination number of first-layer atoms, the plasma frequency of bulk and surface atoms are related by the following equation:
- ,

so that surface and bulk plasmons can be easily distinguished from each other. Plasmon states in a solid are typically localized at the surface, and can strongly affect IMFP.
Vibrational effects
Temperature-dependent atomic lattice vibrations, or phonons, can broaden the core level components and attenuate the interference patterns in an X-ray photoelectron diffraction (XPD) experiment. The simplest way to account for vibrational effects is by multiplying the scattered single-photoelectron wave function by the Debye-Waller factor:

- ,

where is the squared magnitude of the wave vector variation caused by scattering, and is the temperature-dependent one-dimensional vibrational mean squared displacement of the emitter. In the Debye model, the mean squared displacement is calculated in terms of the Debye temperature, , as:





See also
Related methods
- UPS, Ultraviolet photoelectron spectroscopy
- PES, Photoemission spectroscopy
- ZEKE, Zero electron kinetic energy spectroscopy
- AES, Auger electron spectroscopy
- EDS, Energy-dispersive X-ray spectroscopy, (EDX or EDXRF)
- PEEM, Photoelectron emission microscopy
References
- Li, Yang; He, Yongyong; Qiu, Jianxun; Zhao, Jun; Ye, Qianwen; Zhu, Yijie; Mao, Junyuan (2018). "Enhancement of Pitting Corrosion Resistance of Austenitic Stainless Steel Through Deposition of Amorphous/Nanocrystalline Oxy-nitrided Phases by Active Screen Plasma Treatment". Materials Research. 21 (6). doi:10.1590/1980-5373-mr-2017-0697. ISSN 1516-1439.
- Rahmayeni; Alfina, Aimi; Stiadi, Yeni; Lee, Hye Jin; Zulhadjri (2019). "Green synthesis and Characterization of ZnO-CoFe2O4 Semiconductor Photocatalysts Prepared Using Rambutan (Nephelium lappaceum L.) Peel Extract". Materials Research. 22 (5). doi:10.1590/1980-5373-mr-2019-0228. ISSN 1516-1439.
- Gumerova, Nadiia I.; Rompel, Annette (2018-02-07). "Synthesis, structures and applications of electron-rich polyoxometalates". Nature Reviews Chemistry. 2 (2): 1–20. doi:10.1038/s41570-018-0112. ISSN 2397-3358.
- Naumann d'Alnoncourt, Raoul; Csepei, Lénárd-István; Hävecker, Michael; Girgsdies, Frank; Schuster, Manfred E.; Schlögl, Robert; Trunschke, Annette (2014). "The reaction network in propane oxidation over phase-pure MoVTeNb M1 oxide catalysts". Journal of Catalysis. 311: 369–385. doi:10.1016/j.jcat.2013.12.008. hdl:11858/00-001M-0000-0014-F434-5.
- Hävecker, Michael; Wrabetz, Sabine; Kröhnert, Jutta; Csepei, Lenard-Istvan; Naumann d'Alnoncourt, Raoul; Kolen'Ko, Yury V.; Girgsdies, Frank; Schlögl, Robert; Trunschke, Annette (2012). "Surface chemistry of phase-pure M1 MoVTeNb oxide during operation in selective oxidation of propane to acrylic acid". J. Catal. 285: 48–60. doi:10.1016/j.jcat.2011.09.012. hdl:11858/00-001M-0000-0012-1BEB-F.
- Voiry, Damien; Shin, Hyeon Suk; Loh, Kian Ping; Chhowalla, Manish (January 2018). "Low-dimensional catalysts for hydrogen evolution and CO2 reduction". Nature Reviews Chemistry. 2 (1): 0105. doi:10.1038/s41570-017-0105. ISSN 2397-3358.
- Ray, S. and A.G. Shard, Quantitative Analysis of Adsorbed Proteins by X-ray Photoelectron Spectroscopy. Analytical Chemistry, 2011. 83(22): p. 8659-8666.
- Vashishtha, Nitesh; Sapate, Sanjay; Vashishtha, Nitesh; Sapate, Sanjay (2019). "Effect of Experimental Parameters on Wear Response of Thermally Sprayed Carbide Based Coatings". Materials Research. 22 (1). doi:10.1590/1980-5373-mr-2018-0475. ISSN 1516-1439.
- Siegbahn, K.; Edvarson, K. I. Al (1956). "β-Ray spectroscopy in the precision range of 1 : 105". Nuclear Physics. 1 (8): 137–159. Bibcode:1956NucPh...1..137S. doi:10.1016/S0029-5582(56)80022-9.
- Electron Spectroscopy for Atoms, Molecules and Condensed Matter, Nobel Lecture, December 8, 1981
- Turner, D. W.; Jobory, M. I. Al (1962). "Determination of Ionization Potentials by Photoelectron Energy Measurement". The Journal of Chemical Physics. 37 (12): 3007. Bibcode:1962JChPh..37.3007T. doi:10.1063/1.1733134.
- "X-Ray Data Booklet". xdb.lbl.gov. Retrieved 2020-06-20.
- "Handbook of The Elements and Native Oxides" (PDF). XPS International, Inc. Retrieved 8 December 2012.
- Hüfner, S. (1995). Photoelectron spectroscopy: principles and applications. Springer Verlag.
- Sakurai, J. (1995). Modern Quantum Mechanics (Rev. ed.). Addison-Wesley Publishing Company. p. 332. ISBN 0-201-53929-2.
- Attard, Gary; Barnes, Colin (1998). Surfaces. Oxford Chemistry Primers. p. 27. ISBN 978-0198556862.CS1 maint: ref=harv (link)
- "XPS: The Mean Free Path". lasurface.com.
Further reading
- XPS Spectra, Databases, Spectra and Application Notes,
- Handbooks of Monochromatic XPS Spectra - Fully Annotated, PDF of Volumes 1 and 2, B.V.Crist, published by XPS International LLC, 2005, Mountain View, CA, USA
- Handbooks of Monochromatic XPS Spectra, Volumes 1-5, B.V.Crist, published by XPS International LLC, 2004, Mountain View, CA, USA
- Surface Analysis by Auger and X-ray Photoelectron Spectroscopy, ed. J.T.Grant and D.Briggs, published by IM Publications, 2003, Chichester, UK
- An Introduction to Surface Analysis by XPS and AES, J.F.Watts, J.Wolstenholme, published by Wiley & Sons, 2003, Chichester, UK, ISBN 978-0-470-84713-8
- Practical Surface Analysis by Auger and X-ray Photoelectron Spectroscopy, 2nd edition, ed. M.P.Seah and D.Briggs, published by Wiley & Sons, 1992, Chichester, UK
- Practical Surface Analysis by Auger and X-ray Photoelectron Spectroscopy, ed. M.P.Seah and D.Briggs, published by Wiley & Sons, 1983, Chichester, UK ISBN 0-471-26279-X
- Surface Chemical Analysis — Vocabulary, ISO 18115 : 2001, International Organisation for Standardisation (ISO), TC/201, Switzerland,
- Handbook of X-ray Photoelectron Spectroscopy, J.F.Moulder, W.F.Stickle, P.E.Sobol, and K.D.Bomben, published by Perkin-Elmer Corp., 1992, Eden Prairie, MN, USA
External links
- XPS Spectra, Databases, Spectra and Application Notes
- X-Ray Photoelectron Spectroscopy Overview
- The conventional X-Ray source at the Surface Science Laboratory - Instrumental description and guided tour
- The SuperESCA beamline @ Elettra Welcome to the Fast XPS beamline!
- Monochromated XPS Technique background information, useful analysis resources and monochromated XPS equipment description.