Strengthening mechanisms of materials
Methods have been devised to modify the yield strength, ductility, and toughness of both crystalline and amorphous materials. These strengthening mechanisms give engineers the ability to tailor the mechanical properties of materials to suit a variety of different applications. For example, the favorable properties of steel result from interstitial incorporation of carbon into the iron lattice. Brass, a binary alloy of copper and zinc, has superior mechanical properties compared to its constituent metals due to solution strengthening. Work hardening (such as beating a red-hot piece of metal on anvil) has also been used for centuries by blacksmiths to introduce dislocations into materials, increasing their yield strengths.
Basic description
Plastic deformation occurs when large numbers of dislocations move and multiply so as to result in macroscopic deformation. In other words, it is the movement of dislocations in the material which allows for deformation. If we want to enhance a material's mechanical properties (i.e. increase the yield and tensile strength), we simply need to introduce a mechanism which prohibits the mobility of these dislocations. Whatever the mechanism may be, (work hardening, grain size reduction, etc.) they all hinder dislocation motion and render the material stronger than previously.[1][2][3][4]
The stress required to cause dislocation motion is orders of magnitude lower than the theoretical stress required to shift an entire plane of atoms, so this mode of stress relief is energetically favorable. Hence, the hardness and strength (both yield and tensile) critically depend on the ease with which dislocations move. Pinning points, or locations in the crystal that oppose the motion of dislocations,[5] can be introduced into the lattice to reduce dislocation mobility, thereby increasing mechanical strength. Dislocations may be pinned due to stress field interactions with other dislocations and solute particles, creating physical barriers from second phase precipitates forming along grain boundaries. There are five main strengthening mechanisms for metals, each is a method to prevent dislocation motion and propagation, or make it energetically unfavorable for the dislocation to move. For a material that has been strengthened, by some processing method, the amount of force required to start irreversible (plastic) deformation is greater than it was for the original material.
In amorphous materials such as polymers, amorphous ceramics (glass), and amorphous metals, the lack of long range order leads to yielding via mechanisms such as brittle fracture, crazing, and shear band formation. In these systems, strengthening mechanisms do not involve dislocations, but rather consist of modifications to the chemical structure and processing of the constituent material.
The strength of materials cannot infinitely increase. Each of the mechanisms explained below involves some trade-off by which other material properties are compromised in the process of strengthening.
Strengthening mechanisms in metals
Work hardening
The primary species responsible for work hardening are dislocations. Dislocations interact with each other by generating stress fields in the material. The interaction between the stress fields of dislocations can impede dislocation motion by repulsive or attractive interactions. Additionally, if two dislocations cross, dislocation line entanglement occurs, causing the formation of a jog which opposes dislocation motion. These entanglements and jogs act as pinning points, which oppose dislocation motion. As both of these processes are more likely to occur when more dislocations are present, there is a correlation between dislocation density and shear strength.
The shear strengthening provided by dislocation interactions can be described by:[6]

where is a proportionality constant, is the shear modulus, is the Burgers vector, and is the dislocation density.




Dislocation density is defined as the dislocation line length per unit volume:

Similarly, the axial strengthening will be proportional to the dislocation density.

This relationship does not apply when dislocations form cell structures. When cell structures are formed, the average cell size controls the strengthening effect.[6]
Increasing the dislocation density increases the yield strength which results in a higher shear stress required to move the dislocations. This process is easily observed while working a material (by a process of cold working in metals). Theoretically, the strength of a material with no dislocations will be extremely high () because plastic deformation would require the breaking of many bonds simultaneously. However, at moderate dislocation density values of around 107-109 dislocations/m2, the material will exhibit a significantly lower mechanical strength. Analogously, it is easier to move a rubber rug across a surface by propagating a small ripple through it than by dragging the whole rug. At dislocation densities of 1014 dislocations/m2 or higher, the strength of the material becomes high once again. Also, the dislocation density cannot be infinitely high, because then the material would lose its crystalline structure.Citation needed



Solid solution strengthening and alloying
For this strengthening mechanism, solute atoms of one element are added to another, resulting in either substitutional or interstitial point defects in the crystal (see Figure 1). The solute atoms cause lattice distortions that impede dislocation motion, increasing the yield stress of the material. Solute atoms have stress fields around them which can interact with those of dislocations. The presence of solute atoms impart compressive or tensile stresses to the lattice, depending on solute size, which interfere with nearby dislocations, causing the solute atoms to act as potential barriers.
The shear stress required to move dislocations in a material is:

where is the solute concentration and is the strain on the material caused by the solute.


Increasing the concentration of the solute atoms will increase the yield strength of a material, but there is a limit to the amount of solute that can be added, and one should look at the phase diagram for the material and the alloy to make sure that a second phase is not created.
In general, the solid solution strengthening depends on the concentration of the solute atoms, shear modulus of the solute atoms, size of solute atoms, valency of solute atoms (for ionic materials), and the symmetry of the solute stress field. The magnitude of strengthening is higher for non-symmetric stress fields because these solutes can interact with both edge and screw dislocations, whereas symmetric stress fields, which cause only volume change and not shape change, can only interact with edge dislocations.
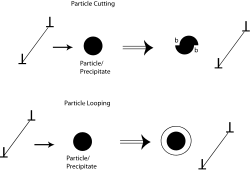
Precipitation hardening
In most binary systems, alloying above a concentration given by the phase diagram will cause the formation of a second phase. A second phase can also be created by mechanical or thermal treatments. The particles that compose the second phase precipitates act as pinning points in a similar manner to solutes, though the particles are not necessarily single atoms.
The dislocations in a material can interact with the precipitate atoms in one of two ways (see Figure 2). If the precipitate atoms are small, the dislocations would cut through them. As a result, new surfaces (b in Figure 2) of the particle would get exposed to the matrix and the particle-matrix interfacial energy would increase. For larger precipitate particles, looping or bowing of the dislocations would occur and result in dislocations getting longer. Hence, at a critical radius of about 5 nm, dislocations will preferably cut across the obstacle, while for a radius of 30 nm, the dislocations will readily bow or loop to overcome the obstacle.
The mathematical descriptions are as follows:
For particle bowing-

For particle cutting-

Dispersion strengthening
Dispersion strengthening is a type of particulate strengthening in which incoherent precipitates attract and pin dislocations. These particles are typically larger than those in the Orowon precipitation hardening discussed above. The effect of dispersion strengthening is effective at high temperatures whereas precipitation strengthening from heat treatments are typically limited to temperatures much lower than the melting temperature of the material.[7] One common type of dispersion strengthening is oxide dispersion strengthening.
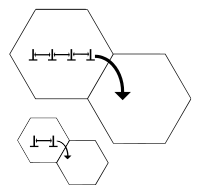
Grain boundary strengthening
In a polycrystalline metal, grain size has a tremendous influence on the mechanical properties. Because grains usually have varying crystallographic orientations, grain boundaries arise. While undergoing deformation, slip motion will take place. Grain boundaries act as an impediment to dislocation motion for the following two reasons:
1. Dislocation must change its direction of motion due to the differing orientation of grains.[4]
2. Discontinuity of slip planes from grain one to grain two.[4]
The stress required to move a dislocation from one grain to another in order to plastically deform a material depends on the grain size. The average number of dislocations per grain decreases with average grain size (see Figure 3). A lower number of dislocations per grain results in a lower dislocation 'pressure' building up at grain boundaries. This makes it more difficult for dislocations to move into adjacent grains. This relationship is the Hall-Petch relationship and can be mathematically described as follows:
,

where is a constant, is the average grain diameter and is the original yield stress.



The fact that the yield strength increases with decreasing grain size is accompanied by the caveat that the grain size cannot be decreased infinitely. As the grain size decreases, more free volume is generated resulting in lattice mismatch. Below approximately 10 nm, the grain boundaries will tend to slide instead; a phenomenon known as grain-boundary sliding. If the grain size gets too small, it becomes more difficult to fit the dislocations in the grain and the stress required to move them is less. It was not possible to produce materials with grain sizes below 10 nm until recently, so the discovery that strength decreases below a critical grain size is still finding new applications.
Transformation hardening
This method of hardening is used for steels.
High-strength steels generally fall into three basic categories, classified by the strengthening mechanism employed. 1- solid-solution-strengthened steels (rephos steels) 2- grain-refined steels or high strength low alloy steels (HSLA) 3- transformation-hardened steels
Transformation-hardened steels are the third type of high-strength steels. These steels use predominantly higher levels of C and Mn along with heat treatment to increase strength. The finished product will have a duplex micro-structure of ferrite with varying levels of degenerate martensite. This allows for varying levels of strength. There are three basic types of transformation-hardened steels. These are dual-phase (DP), transformation-induced plasticity (TRIP), and martensitic steels.
The annealing process for dual -phase steels consists of first holding the steel in the alpha + gamma temperature region for a set period of time. During that time C and Mn diffuse into the austenite leaving a ferrite of greater purity. The steel is then quenched so that the austenite is transformed into martensite, and the ferrite remains on cooling. The steel is then subjected to a temper cycle to allow some level of marten-site decomposition. By controlling the amount of martensite in the steel, as well as the degree of temper, the strength level can be controlled. Depending on processing and chemistry, the strength level can range from 350 to 960 MPa.
TRIP steels also use C and Mn, along with heat treatment, in order to retain small amounts of austenite and bainite in a ferrite matrix. Thermal processing for TRIP steels again involves annealing the steel in the a + g region for a period of time sufficient to allow C and Mn to diffuse into austenite. The steel is then quenched to a point above the martensite start temperature and held there. This allows the formation of bainite, an austenite decomposition product. While at this temperature, more C is allowed to enrich the retained austenite. This, in turn, lowers the martensite start temperature to below room temperature. Upon final quenching a metastable austenite is retained in the predominantly ferrite matrix along with small amounts of bainite (and other forms of decomposed austenite). This combination of micro-structures has the added benefits of higher strengths and resistance to necking during forming. This offers great improvements in formability over other high-strength steels. Essentially, as the TRIP steel is being formed, it becomes much stronger. Tensile strengths of TRIP steels are in the range of 600-960 MPa.
Martensitic steels are also high in C and Mn. These are fully quenched to martensite during processing. The martensite structure is then tempered back to the appropriate strength level, adding toughness to the steel. Tensile strengths for these steels range as high as 1500 MPa.
Strengthening mechanisms in amorphous materials
Polymer
Polymers fracture via breaking of inter- and intra molecular bonds; hence, the chemical structure of these materials plays a huge role in increasing strength. For polymers consisting of chains which easily slide past each other, chemical and physical cross linking can be used to increase rigidity and yield strength. In thermoset polymers (thermosetting plastic), disulfide bridges and other covalent cross links give rise to a hard structure which can withstand very high temperatures. These cross-links are particularly helpful in improving tensile strength of materials which contain lots of free volume prone to crazing, typically glassy brittle polymers.[8] In thermoplastic elastomer, phase separation of dissimilar monomer components leads to association of hard domains within a sea of soft phase, yielding a physical structure with increased strength and rigidity. If yielding occurs by chains sliding past each other (shear bands), the strength can also be increased by introducing kinks into the polymer chains via unsaturated carbon-carbon bonds.[8]
Adding filler materials such as fibers, platelets, and particles is a commonly employed technique for strengthening polymer materials. Fillers such as clay, silica, and carbon network materials have been extensively researched and used in polymer composites in part due to their effect on mechanical properties. Stiffness-confinement effects near rigid interfaces, such as those between a polymer matrix and stiffer filler materials, enhance the stiffness of composites by restricting polymer chain motion.[9] This is especially present where fillers are chemically treated to strongly interact with polymer chains, increasing the anchoring of polymer chains to the filler interfaces and thus further restricting the motion of chains away from the interface.[10] Stiffness-confinement effects have been characterized in model nanocomposites, and shows that composites with length scales on the order of nanometers increase the effect of the fillers on polymer stiffness dramatically.[11]
Increasing the bulkiness of the monomer unit via incorporation of aryl rings is another strengthening mechanism. The anisotropy of the molecular structure means that these mechanisms are heavily dependent on the direction of applied stress. While aryl rings drastically increase rigidity along the direction of the chain, these materials may still be brittle in perpendicular directions. Macroscopic structure can be adjusted to compensate for this anisotropy. For example, the high strength of Kevlar arises from a stacked multilayer macrostructure where aromatic polymer layers are rotated with respect to their neighbors. When loaded oblique to the chain direction, ductile polymers with flexible linkages, such as oriented polyethylene, are highly prone to shear band formation, so macroscopic structures which place the load parallel to the draw direction would increase strength.[8]
Mixing polymers is another method of increasing strength, particularly with materials that show crazing preceding brittle fracture such as atactic polystyrene (APS). For example, by forming a 50/50 mixture of APS with polyphenylene oxide (PPO), this embrittling tendency can be almost completely suppressed, substantially increasing the fracture strength.[8]
Interpenetrating polymer networks (IPNs), consisting of interlacing crosslinked polymer networks that are not covalently bonded to one another, can lead to enhanced strength in polymer materials. The use of an IPN approach imposes compatibility (and thus macroscale homogeneity) on otherwise immiscible blends, allowing for a blending of mechanical properties. For example, silicone-polyurethane IPNs show increased tear and flexural strength over base silicone networks, while preserving the high elastic recovery of the silicone network at high strains.[12] Increased stiffness can also be achieved by pre-straining polymer networks and then sequentially forming a secondary network within the strained material. This takes advantage of the anisotropic strain hardening of the original network (chain alignment from stretching of the polymer chains) and provides a mechanism whereby the two networks transfer stress to one another due to the imposed strain on the pre-strained network.[13]
Glass
Many silicate glasses are strong in compression but weak in tension. By introducing compression stress into the structure, the tensile strength of the material can be increased. This is typically done via two mechanisms: thermal treatment (tempering) or chemical bath (via ion exchange).
In tempered glasses, air jets are used to rapidly cool the top and bottom surfaces of a softened (hot) slab of glass. Since the surface cools quicker, there is more free volume at the surface than in the bulk melt. The core of the slab then pulls the surface inward, resulting in an internal compressive stress at the surface. This substantially increases the tensile strength of the material as tensile stresses exerted on the glass must now resolve the compressive stresses before yielding.

Alternately, in chemical treatment, a glass slab treated containing network formers and modifiers is submerged into a molten salt bath containing ions larger than those present in the modifier. Due to a concentration gradient of the ions, mass transport must take place. As the larger cation diffuses from the molten salt into the surface, it replaces the smaller ion from the modifier. The larger ion squeezing into surface introduces compressive stress in the glass's surface. A common example is treatment of sodium oxide modified silicate glass in molten potassium chloride. Examples of chemically strengthened glass are Gorilla Glass developed and manufactured by Corning, AGC Inc.'s Dragontrail and Schott AG's Xensation.
Composite strengthening
Many of the basic strengthening mechanisms can be classified based on their dimensionality. At 0-D there is precipitate and solid solution strengthening with particulates strengthening structure, at 1-D there is work/forest hardening with line dislocations as the hardening mechanism, and at 2-D there is grain boundary strengthening with surface energy of granular interfaces providing strength improvement. The two primary types of composite strengthening, fiber reinforcement and laminar reinforcement, fall in the 1-D and 2-D classes, respectively. The anisotropy of fiber and laminar composite strength reflects these dimensionalities. The primary idea behind composite strengthening is to combine materials with opposite strengths and weaknesses to create a material which transfers load onto the stiffer material but benefits from the ductility and toughness of the softer material.[14]
Fiber reinforcement
Fiber-reinforced composites (FRCs) consist of a matrix of one material containing parallel embedded fibers. There are two variants of fiber-reinforced composites, one with stiff fibers and a ductile matrix and one with ductile fibers and a stiff matrix. The former variant is exemplified by fiberglass which contains very strong but delicate glass fibers embedded in a softer plastic matrix resilient to fracture. The latter variant is found in almost all buildings as reinforced concrete with ductile, high tensile-strength steel rods embedded in brittle, high compressive-strength concrete. In both cases, the matrix and fibers have complimentary mechanical properties and the resulting composite material is therefore more practical for applications in the real world.
For a composite containing aligned, stiff fibers which span the length of the material and a soft, ductile matrix, the following descriptions provide a rough model.
Four stages of deformation
The condition of a fiber-reinforced composite under applied tensile stress along the direction of the fibers can be decomposed into four stages from small strain to large strain. Since the stress is parallel to the fibers, the deformation is described by the isostrain condition, i.e., the fiber and matrix experience the same strain. At each stage, the composite stress () is given in terms of the volume fractions of the fiber and matrix (), the Young's moduli of the fiber and matrix (), the strain of the composite (), and the stress of the fiber and matrix as read from a stress-strain curve ().





- Both fiber and composite remain in the elastic strain regime. In this stage, we also note that the composite Young's modulus is a simple weighted sum of the two component moduli.
- The fiber remains in the elastic regime but the matrix yields and plastically deforms.
- Both fiber and composite yield and plastically deform. This stage often features significant Poisson strain which is not captured by model below.
- The fiber fractures while the matrix continues to plastically deform. While in reality the fractured pieces of fiber still contribute some strength, it is left out of this simple model.





Tensile strength
Due to the heterogeneous nature of FRCs, they also feature multiple tensile strengths (TS), one corresponding to each component. Given the assumptions outlined above, the first tensile strength would correspond to failure of the fibers, with some support from the matrix plastic deformation strength, and the second with failure of the matrix.


Anisotropy (Orientation effects)
As a result of the aforementioned dimensionality (1-D) of fiber reinforcement, significant anisotropy is observed in its mechanical properties. The following equations model the tensile strength of a FRC as a function of the misalignment angle () between the fibers and the applied force, the stresses in the parallel and perpendicular, or and o, cases (), and the shear strength of the matrix ().





- Small Misalignment Angle (longitudinal fracture)
- The angle is small enough to maintain load transfer onto the fibers and prevent delamination of fibers and the misaligned stress samples a slightly larger cross-sectional area of the fiber so the strength of the fiber is not just maintained but actually increases compared to the parallel case.
- [14]

- Significant Misalignment Angle (shear failure)
- The angle is large enough that the load is not effectively transferred to the fibers and the matrix experiences enough strain to fracture.
- [14]

- Near Perpendicular Misaligment Angle (transverse fracture)
- The angle is close to 90o so most of the load remains in the matrix and thus tensile transverse matrix fracture is the dominant failure condition. This can be seen as complementary to the small angle case, with similar form but with an angle .
- [14]


Laminar reinforcement
Applications and current research
Strengthening of materials is useful in many applications. A primary application of strengthened materials is for construction. In order to have stronger buildings and bridges, one must have a strong frame that can support high tensile or compressive load and resist plastic deformation. The steel frame used to make the building should be as strong as possible so that it does not bend under the entire weight of the building. Polymeric roofing materials would also need to be strong so that the roof does not cave in when there is build-up of snow on the rooftop.
Research is also currently being done to increase the strength of metallic materials through the addition of polymer materials such as bonded carbon fiber reinforced polymer to (CFRP).
Molecular dynamics simulations
The use of computation simulations to model work hardening in materials allows for the direct observation of critical elements that rule the process of strengthening materials. The basic reasoning derives from the fact that, when examining plasticity and the movement of dislocations in materials, a focus on the atomistic level is many times not accounted for and the focus rests on the contiuum description of materials. Since the practice of tracking these atomistic effects in experiments and theorizing about them in textbooks cannot provide a full understanding of these interactions, many turn to molecular dynamics simulations to develop this understanding.[15]
The simulations work by utilizing the known atomic interactions between any two atoms and the relationship F = ma, so that the dislocations moving through the material are ruled by simple mechanical actions and reactions of the atoms. The interatomic potential usually utilized to estimate these interactions is the Lennard – Jones 12:6 potential. Lennard – Jones is widely accepted because its experimental shortcomings are well-known.[15][16] These interactions are simply scaled up to millions or billions of atoms in some cases to simulate materials more accurately.
Molecular dynamic simulations display the interactions based upon the governing equations provided above for the strengthening mechanisms. They provide an effective way to see these mechanisms in action outside the painstaking realm of direct observation during experiments.
See also
- Grain boundary strengthening
- Precipitation strengthening
- Solid solution strengthening
- Strength of materials
- Tempering (metallurgy)
- Work hardening
References
- Davidge, R.W., Mechanical Behavior of Ceramics, Cambridge Solid State Science Series, (1979)
- Lawn, B.R., Fracture of Brittle Solids, Cambridge Solid State Science Series, 2nd Edn. (1993)
- Green, D., An Introduction to the Mechanical Properties of Ceramics, Cambridge Solid State Science Series, Eds. Clarke, D.R., Suresh, S., Ward, I.M. (1998)
- Callister, William Jr, Materials Science and Engineering, An Introduction. John Wiley & Sons, NY, NY (1985)
- Kuhlmann-Wilsdorf, D., "Theory of Plastic Deformation," Materials Science and Engineering A, vol 113, pp 1-42, July 1989
- Soboyejo, Wole O. (2003). "8.4 Dislocation Strengthening". Mechanical properties of engineered materials. Marcel Dekker. ISBN 0-8247-8900-8. OCLC 300921090.
- Soboyejo, Wole O. (2003). "8.7 Dispersion Strengthening". Mechanical properties of engineered materials. Marcel Dekker. ISBN 0-203-91039-7. OCLC 54091550.
- Meyers, Chawla. Mechanical Behavior of Materials. Cambridge University Press. pg 420-425. 1999
- Krutyeva, M.; Wischnewski, A.; Monkenbusch, M.; Willner, L.; Maiz, J.; Mijangos, C.; Arbe, A.; Colmenero, J.; Radulescu, A.; Holderer, O.; Ohl, M.; Richter, D. (2013). "Effect of Nanoconfinement on Polymer Dynamics: Surface Layers and Interphases". Physical Review Letters. 110 (10): 108303. doi:10.1103/PhysRevLett.110.108303. hdl:10261/102483. PMID 23521308.
- Ligon-Auer, Samuel Clark; Schwentenwein, Martin; Gorsche, Christian; Stampfl, Jürgen; Liska, Robert (2016). "Toughening of photo-curable polymer networks: A review". Polymer Chemistry. 7 (2): 257–286. doi:10.1039/C5PY01631B.
- Zhang, Min; Askar, Shadid; Torkelson, John M.; Brinson, L. Catherine (2017). "Stiffness Gradients in Glassy Polymer Model Nanocomposites: Comparisons of Quantitative Characterization by Fluorescence Spectroscopy and Atomic Force Microscopy". Macromolecules. 50 (14): 5447–5458. doi:10.1021/acs.macromol.7b00917.
- Arkles, Barry; Crosby, Jane (1989). "Polysiloxane—Thermoplastic Interpenetrating Polymer Networks". Silicon-Based Polymer Science. Advances in Chemistry. 224. pp. 181–199. doi:10.1021/ba-1990-0224.ch010. ISBN 0-8412-1546-4.
- Singh, Naveen K.; Lesser, Alan J. (2010). "Mechanical and thermo-mechanical studies of double networks based on thermoplastic elastomers". Journal of Polymer Science Part B: Polymer Physics. 48 (7): 778–789. doi:10.1002/polb.21943.
- Courtney, Thomas H (1990). Mechanical Behavior of Materials (2nd ed.). Long Grove, IL: Waveland Press Inc.
- Buehler, Markus J. the dynamical complexity of work - hardening: a large-scale molecular dynamics simulation. Acta Mech Sinica. pg 103-111. 2005
- Abraham, F. "Simulating materials failure by using up to one billion atoms and the world's fastest computer: Work-hardening." Proc Natl Acad Sci. pg 5783–5787. 2002