Protein–protein interaction prediction
Protein–protein interaction prediction is a field combining bioinformatics and structural biology in an attempt to identify and catalog physical interactions between pairs or groups of proteins. Understanding protein–protein interactions is important for the investigation of intracellular signaling pathways, modelling of protein complex structures and for gaining insights into various biochemical processes.
Experimentally, physical interactions between pairs of proteins can be inferred from a variety of techniques, including yeast two-hybrid systems, protein-fragment complementation assays (PCA), affinity purification/mass spectrometry, protein microarrays, fluorescence resonance energy transfer (FRET), and Microscale Thermophoresis (MST). Efforts to experimentally determine the interactome of numerous species are ongoing. Experimentally determined interactions usually provide the basis for computational methods to predict interactions, e.g. using homologous protein sequences across species. However, there are also methods that predict interactions de novo, without prior knowledge of existing interactions.
Methods
Proteins that interact are more likely to co-evolve,[1][2][3][4] therefore, it is possible to make inferences about interactions between pairs of proteins based on their phylogenetic distances. It has also been observed in some cases that pairs of interacting proteins have fused orthologues in other organisms. In addition, a number of bound protein complexes have been structurally solved and can be used to identify the residues that mediate the interaction so that similar motifs can be located in other organisms.
Phylogenetic profiling
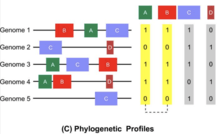
The phylogenetic profile method is based on the hypothesis that if two or more proteins are concurrently present or absent across several genomes, then they are likely functionally related.[5] Figure A illustrates a hypothetical situation in which proteins A and B are identified as functionally linked due to their identical phylogenetic profiles across 5 different genomes. The Joint Genome Institute provides an Integrated Microbial Genomes and Microbiomes database (JGI IMG) that has a phylogenetic profiling tool for single genes and gene cassettes.
Prediction of co-evolved protein pairs based on similar phylogenetic trees
It was observed that the phylogenetic trees of ligands and receptors were often more similar than due to random chance.[4] This is likely because they faced similar selection pressures and co-evolved. This method[6] uses the phylogenetic trees of protein pairs to determine if interactions exist. To do this, homologs of the proteins of interest are found (using a sequence search tool such as BLAST) and multiple-sequence alignments are done (with alignment tools such as Clustal) to build distance matrices for each of the proteins of interest.[4] The distance matrices should then be used to build phylogenetic trees. However, comparisons between phylogenetic trees are difficult, and current methods circumvent this by simply comparing distance matrices[4]. The distance matrices of the proteins are used to calculate a correlation coefficient, in which a larger value corresponds to co-evolution. The benefit of comparing distance matrices instead of phylogenetic trees is that the results do not depend on the method of tree building that was used. The downside is that difference matrices are not perfect representations of phylogenetic trees, and inaccuracies may result from using such a shortcut.[4] Another factor worthy of note is that there are background similarities between the phylogenetic trees of any protein, even ones that do not interact. If left unaccounted for, this could lead to a high false-positive rate. For this reason, certain methods construct a background tree using 16S rRNA sequences which they use as the canonical tree of life. The distance matrix constructed from this tree of life is then subtracted from the distance matrices of the proteins of interest.[7] However, because RNA distance matrices and DNA distance matrices have different scale, presumably because RNA and DNA have different mutation rates, the RNA matrix needs to be rescaled before it can be subtracted from the DNA matrices.[7] By using molecular clock proteins, the scaling coefficient for protein distance/RNA distance can be calculated.[7] This coefficient is used to rescale the RNA matrix.
Rosetta stone (gene fusion) method
The Rosetta Stone or Domain Fusion method is based on the hypothesis that interacting proteins are sometimes fused into a single protein[3]. For instance, two or more separate proteins in a genome may be identified as fused into one single protein in another genome. The separate proteins are likely to interact and thus are likely functionally related. An example of this is the Human Succinyl coA Transferase enzyme, which is found as one protein in humans but as two separate proteins, Acetate coA Transferase alpha and Acetate coA Transferase beta, in Escherichia coli[3]. In order to identify these sequences, a sequence similarity algorithm such as the one used by BLAST is necessary. For example, if we had the amino acid sequences of proteins A and B and the amino acid sequences of all proteins in a certain genome, we could check each protein in that genome for non-overlapping regions of sequence similarity to both proteins A and B. Figure B depicts the BLAST sequence alignment of Succinyl coA Transferase with its two separate homologs in E. coli. The two subunits have non-overlapping regions of sequence similarity with the human protein, indicated by the pink regions, with the alpha subunit similar to the first half of the protein and the beta similar to the second half. One limit of this method is that not all proteins that interact can be found fused in another genome, and therefore cannot be identified by this method. On the other hand, the fusion of two proteins does not necessitate that they physically interact. For instance, the SH2 and SH3 domains in the src protein are known to interact. However, many proteins possess homologs of these domains and they do not all interact[3].
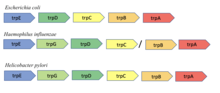
Conserved gene neighborhood
The conserved neighborhood method is based on the hypothesis that if genes encoding two proteins are neighbors on a chromosome in many genomes, then they are likely functionally related. The method is based on an observation by Bork et al. of gene pair conservation across nine bacterial and archaeal genomes. The method is most effective in prokaryotes with operons as the organization of genes in an operon is generally related to function[8]. For instance, the trpA and trpB genes in Escherichia coli encode the two subunits of the tryptophan synthase enzyme known to interact to catalyze a single reaction. The adjacency of these two genes was shown to be conserved across nine different bacterial and archaeal genomes[8].
Classification methods
Classification methods use data to train a program (classifier) to distinguish positive examples of interacting protein/domain pairs with negative examples of non-interacting pairs. Popular classifiers used are Random Forest Decision (RFD) and Support Vector Machines. RFD produces results based on the domain composition of interacting and non-interacting protein pairs. When given a protein pair to classify, RFD first creates a representation of the protein pair in a vector.[9] The vector contains all the domain types used to train RFD, and for each domain type the vector also contains a value of 0, 1, or 2. If the protein pair does not contain a certain domain, then the value for that domain is 0. If one of the proteins of the pair contains the domain, then the value is 1. If both proteins contain the domain, then the value is 2.[9] Using training data, RFD constructs a decision forest, consisting of many decision trees. Each decision tree evaluates several domains, and based on the presence or absence of interactions in these domains, makes a decision as to if the protein pair interacts. The vector representation of the protein pair is evaluated by each tree to determine if they are an interacting pair or a non-interacting pair. The forest tallies up all the input from the trees to come up with a final decision.[9] The strength of this method is that it does not assume that domains interact independent of each other. This makes it so that multiple domains in proteins can be used in the prediction.[9] This is a big step up from previous methods which could only predict based on a single domain pair. The limitation of this method is that it relies on the training dataset to produce results. Thus, usage of different training datasets could influence the results.
Inference of interactions from homologous structures
This group of methods[10][9][11][12][13][14] makes use of known protein complex structures to predict and structurally model interactions between query protein sequences. The prediction process generally starts by employing a sequence based method (e.g. Interolog) to search for protein complex structures that are homologous to the query sequences. These known complex structures are then used as templates to structurally model the interaction between query sequences. This method has the advantage of not only inferring protein interactions but also suggests models of how proteins interact structurally, which can provide some insights into the atomic level mechanism of that interaction. On the other hand, the ability for these methods to make a prediction is constrained by a limited number of known protein complex structures.
Association methods
Association methods look for characteristic sequences or motifs that can help distinguish between interacting and non-interacting pairs. A classifier is trained by looking for sequence-signature pairs where one protein contains one sequence-signature, and its interacting partner contains another sequence-signature.[15] They look specifically for sequence-signatures that are found together more often than by chance. This uses a log-odds score which is computed as log2(Pij/PiPj), where Pij is the observed frequency of domains i and j occurring in one protein pair; Pi and Pj are the background frequencies of domains i and j in the data. Predicted domain interactions are those with positive log-odds scores and also having several occurrences within the database.[15] The downside with this method is that it looks at each pair of interacting domains separately, and it assumes that they interact independently of each other.
Identification of structural patterns
This method[16][17] builds a library of known protein–protein interfaces from the PDB, where the interfaces are defined as pairs of polypeptide fragments that are below a threshold slightly larger than the Van der Waals radius of the atoms involved. The sequences in the library are then clustered based on structural alignment and redundant sequences are eliminated. The residues that have a high (generally >50%) level of frequency for a given position are considered hotspots.[18] This library is then used to identify potential interactions between pairs of targets, providing that they have a known structure (i.e. present in the PDB).
Bayesian network modelling
Bayesian methods[19] integrate data from a wide variety of sources, including both experimental results and prior computational predictions, and use these features to assess the likelihood that a particular potential protein interaction is a true positive result. These methods are useful because experimental procedures, particularly the yeast two-hybrid experiments, are extremely noisy and produce many false positives, while the previously mentioned computational methods can only provide circumstantial evidence that a particular pair of proteins might interact.[20]
Domain-pair exclusion analysis
The domain-pair exclusion analysis[21] detects specific domain interactions that are hard to detect using Bayesian methods. Bayesian methods are good at detecting nonspecific promiscuous interactions and not very good at detecting rare specific interactions. The domain-pair exclusion analysis method calculates an E-score which measures if two domains interact. It is calculated as log(probability that the two proteins interact given that the domains interact/probability that the two proteins interact given that the domains don’t interact). The probabilities required in the formula are calculated using an Expectation Maximization procedure, which is a method for estimating parameters in statistical models. High E-scores indicate that the two domains are likely to interact, while low scores indicate that other domains form the protein pair are more likely to be responsible for the interaction. The drawback with this method is that it does not take into account false positives and false negatives in the experimental data.
Supervised learning problem
The problem of PPI prediction can be framed as a supervised learning problem. In this paradigm the known protein interactions supervise the estimation of a function that can predict whether an interaction exists or not between two proteins given data about the proteins (e.g., expression levels of each gene in different experimental conditions, location information, phylogenetic profile, etc.).
Relationship to docking methods
The field of protein–protein interaction prediction is closely related to the field of protein–protein docking, which attempts to use geometric and steric considerations to fit two proteins of known structure into a bound complex. This is a useful mode of inquiry in cases where both proteins in the pair have known structures and are known (or at least strongly suspected) to interact, but since so many proteins do not have experimentally determined structures, sequence-based interaction prediction methods are especially useful in conjunction with experimental studies of an organism's interactome.
See also
- Interactome
- Protein–protein interaction
- Macromolecular docking
- Protein–DNA interaction site predictor
- Two-hybrid screening
- Protein structure prediction software
- FastContact
References
- Dandekar T., Snel B.,Huynen M. and Bork P. (1998) "Conservation of gene order: a fingerprint of proteins that physically interact." Trends Biochem. Sci. (23),324-328
- Enright A.J.,Iliopoulos I.,Kyripides N.C. and Ouzounis C.A. (1999) "Protein interaction maps for complete genomes based on gene fusion events." Nature (402), 86-90
- Marcotte E.M., Pellegrini M., Ng H.L., Rice D.W., Yeates T.O., Eisenberg D. (1999) "Detecting protein function and protein-protein interactions from genome sequences." Science (285), 751-753
- Pazos, F.; Valencia, A. (2001). "Similarity of phylogenetic trees as indicator of protein-protein interaction". Protein Engineering. 9 (14): 609–614. doi:10.1093/protein/14.9.609.
- Raman, Karthik (2010-02-15). "Construction and analysis of protein–protein interaction networks". Automated Experimentation. 2 (1): 2. doi:10.1186/1759-4499-2-2. ISSN 1759-4499. PMC 2834675. PMID 20334628.
- Tan S.H., Zhang Z., Ng S.K. (2004) "ADVICE: Automated Detection and Validation of Interaction by Co-Evolution." Nucl. Ac. Res., 32 (Web Server issue):W69-72.
- Pazos, F; Ranea, JA; Juan, D; Sternberg, MJ (2005). "Assessing protein coevolution in the context of the tree of life assists in the prediction of the interactome". J Mol Biol. 352 (4): 1002–1015. doi:10.1016/j.jmb.2005.07.005. PMID 16139301.
- Dandekar, T. (1998-09-01). "Conservation of gene order: a fingerprint of proteins that physically interact". Trends in Biochemical Sciences. 23 (9): 324–328. doi:10.1016/S0968-0004(98)01274-2. ISSN 0968-0004.
- Chen, XW; Liu, M (2005). "Prediction of protein–protein interactions using random decision forest framework". Bioinformatics. 21 (24): 4394–4400. doi:10.1093/bioinformatics/bti721. PMID 16234318.
- Aloy, P.; Russell, R. B. (2003). "InterPreTS: protein Interaction Prediction through Tertiary Structure". Bioinformatics. 19 (1): 161–162. doi:10.1093/bioinformatics/19.1.161.
- Fukuhara, Naoshi, and Takeshi Kawabata. (2008) "HOMCOS: a server to predict interacting protein pairs and interacting sites by homology modeling of complex structures" Nucleic Acids Research, 36 (S2): 185-.
- Kittichotirat W, M Guerquin, RE Bumgarner, and R Samudrala (2009) "Protinfo PPC: a web server for atomic level prediction of protein complexes" Nucleic Acids Research, 37 (Web Server issue): 519-25.
- Shoemaker, BA; Zhang, D; Thangudu, RR; Tyagi, M; Fong, JH; Marchler-Bauer, A; Bryant, SH; Madej, T; Panchenko, AR (Jan 2010). "Inferred Biomolecular Interaction Server--a web server to analyze and predict protein interacting partners and binding sites". Nucleic Acids Res. 38 (Database issue): D518–24. doi:10.1093/nar/gkp842. PMC 2808861. PMID 19843613.
- Esmaielbeiki, R; Nebel, J-C (2014). "Scoring docking conformations using predicted protein interfaces". BMC Bioinformatics. 15: 171. doi:10.1186/1471-2105-15-171. PMC 4057934. PMID 24906633.
- Sprinzak, E; Margalit, H (2001). "Correlated sequence-signatures as markers of protein–protein interaction". J Mol Biol. 311 (4): 681–692. doi:10.1006/jmbi.2001.4920. PMID 11518523.
- Aytuna, A. S.; Keskin, O.; Gursoy, A. (2005). "Prediction of protein-protein interactions by combining structure and sequence conservation in protein interfaces". Bioinformatics. 21 (12): 2850–2855. doi:10.1093/bioinformatics/bti443. PMID 15855251.
- Ogmen, U.; Keskin, O.; Aytuna, A.S.; Nussinov, R.; Gursoy, A. (2005). "PRISM: protein interactions by structural matching". Nucl. Ac. Res. 33: W331–336. doi:10.1093/nar/gki585.
- Keskin, O.; Ma, B.; Nussinov, R. (2004). "Hot regions int protein-protein interactions: The organization and contribution of structurally conserved hot spot residues". J. Mol. Biol. 345 (5): 1281–1294. doi:10.1016/j.jmb.2004.10.077. PMID 15644221.
- Jansen, R; Yu, H; Greenbaum, D; Kluger, Y; Krogan, NJ; Chung, S; Emili, A; Snyder, M; Greenblatt, JF; Gerstein, M (2003). "A Bayesian networks approach for predicting protein-protein interactions from genomic data". Science. 302 (5644): 449–53. Bibcode:2003Sci...302..449J. CiteSeerX 10.1.1.217.8151. doi:10.1126/science.1087361. PMID 14564010.
- Zhang, QC; Petrey, D; Deng, L; Qiang, L; Shi, Y; Thu, CA; Bisikirska, B; Lefebvre, C; Accili, D; Hunter, T; Maniatis, T; Califano, A; Honig, B (2012). "Structure-based prediction of protein-protein interactions on a genome-wide scale". Nature. 490 (7421): 556–60. Bibcode:2012Natur.490..556Z. doi:10.1038/nature11503. PMC 3482288. PMID 23023127.
- Shoemaker, BA; Panchenko, AR (2007). "Deciphering protein–protein interactions. Part II. Computational methods to predict protein and domain interaction partners". PLoS Comput Biol. 3 (4): e43. Bibcode:2007PLSCB...3...43S. doi:10.1371/journal.pcbi.0030043. PMC 1857810. PMID 17465672.
External links
- Overview of protein interaction databases
- ChiPPI: The Server Protein-Protein Interaction of Chimeric Proteins.