Protein mass spectrometry
Protein mass spectrometry refers to the application of mass spectrometry to the study of proteins. Mass spectrometry is an important method for the accurate mass determination and characterization of proteins, and a variety of methods and instrumentations have been developed for its many uses. Its applications include the identification of proteins and their post-translational modifications, the elucidation of protein complexes, their subunits and functional interactions, as well as the global measurement of proteins in proteomics. It can also be used to localize proteins to the various organelles, and determine the interactions between different proteins as well as with membrane lipids.[1][2]

The two primary methods used for the ionization of protein in mass spectrometry are electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI). These ionization techniques are used in conjunction with mass analyzers such as tandem mass spectrometry. In general, the protein are analyzed either in a "top-down" approach in which proteins are analyzed intact, or a "bottom-up" approach in which protein are first digested into fragments. An intermediate "middle-down" approach in which larger peptide fragments are analyzed may also sometimes be used.
History
The application of mass spectrometry to study proteins became popularized in the 1980s after the development of MALDI and ESI. These ionization techniques have played a significant role in the characterization of proteins. (MALDI) Matrix-assisted laser desorption ionization was coined in the late 80's by Franz Hillenkamp and Michael Karas.[3] Hillenkamp, Karas and their fellow researchers were able to ionize the amino acid alanine by mixing it with the amino acid tryptophan and irradiated with a pulse 266 nm laser.[3] Though important, the breakthrough did not come until 1987. In 1987, Koichi Tanaka used the "ultra fine metal plus liquid matrix method" and ionized biomolecules the size of 34,472 Da protein carboxypeptidase-A.[4]
In 1968, Malcolm Dole reported the first use of electrospray ionization with mass spectrometry. Around the same time MALDI became popularized, John Bennett Fenn was cited for the development of electrospray ionization.[5][6] Koichi Tanaka received the 2002 Nobel Prize in Chemistry alongside John Fenn, and Kurt Wüthrich "for the development of methods for identification and structure analyses of biological macromolecules."[7] These ionization methods have greatly facilitated the study of proteins by mass spectrometry. Consequently, protein mass spectrometry now plays a leading role in protein characterization.
Methods and approaches
Techniques
Mass spectrometry of proteins requires that the proteins in solution or solid state be turned into an ionized form in the gas phase before they are injected and accelerated in an electric or magnetic field for analysis. The two primary methods for ionization of proteins are electrospray ionization (ESI) and matrix-assisted laser desorption/ionization (MALDI). In electrospray, the ions are created from proteins in solution, and it allows fragile molecules to be ionized intact, sometimes preserving non-covalent interactions. In MALDI, the proteins are embedded within a matrix normally in a solid form, and ions are created by pulses of laser light. Electrospray produces more multiply-charged ions than MALDI, allowing for measurement of high mass protein and better fragmentation for identification, while MALDI is fast and less likely to be affected by contaminants, buffers and additives.[8]
Whole-protein mass analysis is primarily conducted using either time-of-flight (TOF) MS, or Fourier transform ion cyclotron resonance (FT-ICR). These two types of instrument are preferable here because of their wide mass range, and in the case of FT-ICR, its high mass accuracy. Electrospray ionization of a protein often results in generation of multiple charged species of 800 < m/z < 2000 and the resultant spectrum can be deconvoluted to determine the protein's average mass to within 50 ppm or better using TOF or ion-trap instruments.
Mass analysis of proteolytic peptides is a popular method of protein characterization, as cheaper instrument designs can be used for characterization. Additionally, sample preparation is easier once whole proteins have been digested into smaller peptide fragments. The most widely used instrument for peptide mass analysis are the MALDI-TOF instruments as they permit the acquisition of peptide mass fingerprints (PMFs) at high pace (1 PMF can be analyzed in approx. 10 sec). Multiple stage quadrupole-time-of-flight and the quadrupole ion trap also find use in this application.
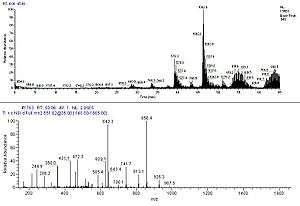
Tandem mass spectrometry (MS/MS) is used to measure fragmentation spectra and identify proteins at high speed and accuracy. Collision-induced dissociation is used in mainstream applications to generate a set of fragments from a specific peptide ion. The fragmentation process primarily gives rise to cleavage products that break along peptide bonds. Because of this simplicity in fragmentation, it is possible to use the observed fragment masses to match with a database of predicted masses for one of many given peptide sequences. Tandem MS of whole protein ions has been investigated recently using electron capture dissociation and has demonstrated extensive sequence information in principle but is not in common practice.
Approaches
In keeping with the performance and mass range of available mass spectrometers, two approaches are used for characterizing proteins. In the first, intact proteins are ionized by either of the two techniques described above, and then introduced to a mass analyzer. This approach is referred to as "top-down" strategy of protein analysis as it involves starting with the whole mass and then pulling it apart. The top-down approach however is mostly limited to low-throughput single-protein studies due to issues involved in handling whole proteins, their heterogeneity and the complexity of their analyses.[8]
In the second approach, referred to as the "bottom-up" MS, proteins are enzymatically digested into smaller peptides using a protease such as trypsin. Subsequently, these peptides are introduced into the mass spectrometer and identified by peptide mass fingerprinting or tandem mass spectrometry. Hence, this approach uses identification at the peptide level to infer the existence of proteins pieced back together with de novo repeat detection.[9] The smaller and more uniform fragments are easier to analyze than intact proteins and can be also determined with high accuracy, this "bottom-up" approach is therefore the preferred method of studies in proteomics. A further approach that is beginning to be useful is the intermediate "middle-down" approach in which proteolytic peptides larger than the typical tryptic peptides are analyzed.[8]
Protein and peptide fractionation

Proteins of interest are usually part of a complex mixture of multiple proteins and molecules, which co-exist in the biological medium. This presents two significant problems. First, the two ionization techniques used for large molecules only work well when the mixture contains roughly equal amounts of material, while in biological samples, different proteins tend to be present in widely differing amounts. If such a mixture is ionized using electrospray or MALDI, the more abundant species have a tendency to "drown" or suppress signals from less abundant ones. Second, mass spectrum from a complex mixture is very difficult to interpret due to the overwhelming number of mixture components. This is exacerbated by the fact that enzymatic digestion of a protein gives rise to a large number of peptide products.
In light of these problems, the methods of one- and two-dimensional gel electrophoresis and high performance liquid chromatography are widely used for separation of proteins. The first method fractionates whole proteins via two-dimensional gel electrophoresis. The first-dimension of 2D gel is isoelectric focusing (IEF). In this dimension, the protein is separated by its isoelectric point (pI) and the second-dimension is SDS-polyacrylamide gel electrophoresis (SDS-PAGE). This dimension separates the protein according to its molecular weight.[10] Once this step is completed in-gel digestion occurs. In some situations, it may be necessary to combine both of these techniques. Gel spots identified on a 2D Gel are usually attributable to one protein. If the identity of the protein is desired, usually the method of in-gel digestion is applied, where the protein spot of interest is excised, and digested proteolytically. The peptide masses resulting from the digestion can be determined by mass spectrometry using peptide mass fingerprinting. If this information does not allow unequivocal identification of the protein, its peptides can be subject to tandem mass spectrometry for de novo sequencing. Small changes in mass and charge can be detected with 2D-PAGE. The disadvantages with this technique are its small dynamic range compared to other methods, some proteins are still difficult to separate due to their acidity, basicity, hydrophobicity, and size (too large or too small).[11]
The second method, high performance liquid chromatography is used to fractionate peptides after enzymatic digestion. Characterization of protein mixtures using HPLC/MS is also called shotgun proteomics and MuDPIT (Multi-Dimensional Protein Identification Technology). A peptide mixture that results from digestion of a protein mixture is fractionated by one or two steps of liquid chromatography. The eluent from the chromatography stage can be either directly introduced to the mass spectrometer through electrospray ionization, or laid down on a series of small spots for later mass analysis using MALDI.
Applications
Protein identification
There are two main ways MS is used to identify proteins. Peptide mass fingerprinting uses the masses of proteolytic peptides as input to a search of a database of predicted masses that would arise from digestion of a list of known proteins. If a protein sequence in the reference list gives rise to a significant number of predicted masses that match the experimental values, there is some evidence that this protein was present in the original sample. Purification steps therefore limit the throughput of the peptide mass fingerprinting approach. Peptide mass fingerprinting can be achieved with MS/MS.[9]
MS is also the preferred method for the identification of post-translational modifications in proteins as it is more advantageous than other approaches such as the antibody-based methods.[1]
De novo (peptide) sequencing
De novo peptide sequencing for mass spectrometry is typically performed without prior knowledge of the amino acid sequence. It is the process of assigning amino acids from peptide fragment masses of a protein. De novo sequencing has proven successful for confirming and expanding upon results from database searches.
As de novo sequencing is based on mass and some amino acids have identical masses (e.g. leucine and isoleucine), accurate manual sequencing can be difficult. Therefore, it may be necessary to utilize a sequence homology search application to work in tandem between a database search and de novo sequencing to address this inherent limitation.
Database searching has the advantage of quickly identifying sequences, provided they have already been documented in a database. Other inherent limitations of database searching include sequence modifications/mutations (some database searches do not adequately account for alterations to the 'documented' sequence, thus can miss valuable information), the unknown (if a sequence is not documented, it will not be found), false positives, and incomplete and corrupted data.[12]
An annotated peptide spectral library can also be used as a reference for protein/peptide identification. It offers the unique strength of reduced search space and increased specificity. The limitations include spectra not included in the library will not be identified, spectra collected from different types of mass spectrometers can have quite distinct features, and reference spectra in the library may contain noise peaks, which may lead to false positive identifications.[13] A number of different algorithmic approaches have been described to identify peptides and proteins from tandem mass spectrometry (MS/MS), peptide de novo sequencing and sequence tag-based searching.[14]
Protein quantitation
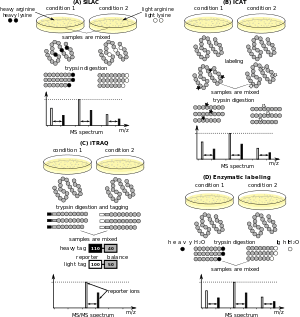
Multiple methods allow for the quantitation of proteins by mass spectrometry (quantitative proteomics),[15] and recent advances have enabled quantifying thousands of proteins in single cells.[16][17] Typically, stable (e.g. non-radioactive) heavier isotopes of carbon (13C) or nitrogen (15N) are incorporated into one sample while the other one is labeled with corresponding light isotopes (e.g. 12C and 14N).[18] The two samples are mixed before the analysis. Peptides derived from the different samples can be distinguished due to their mass difference. The ratio of their peak intensities corresponds to the relative abundance ratio of the peptides (and proteins). The most popular methods for isotope labeling are SILAC (stable isotope labeling by amino acids in cell culture), trypsin-catalyzed 18O labeling, ICAT (isotope coded affinity tagging), iTRAQ (isobaric tags for relative and absolute quantitation).[19] “Semi-quantitative” mass spectrometry can be performed without labeling of samples.[20] Typically, this is done with MALDI analysis (in linear mode). The peak intensity, or the peak area, from individual molecules (typically proteins) is here correlated to the amount of protein in the sample. However, the individual signal depends on the primary structure of the protein, on the complexity of the sample, and on the settings of the instrument. Other types of "label-free" quantitative mass spectrometry, uses the spectral counts (or peptide counts) of digested proteins as a means for determining relative protein amounts.[12]
Protein structure determination
Characteristics indicative of the 3-dimensional structure of proteins can be probed with mass spectrometry in various ways.[21] By using chemical crosslinking to couple parts of the protein that are close in space, but far apart in sequence, information about the overall structure can be inferred. By following the exchange of amide protons with deuterium from the solvent, it is possible to probe the solvent accessibility of various parts of the protein.[22] Hydrogen-deuterium exchange mass spectrometry has been used to study proteins and their conformations for over 20 years. This type of protein structural analysis can be suitable for proteins that are challenging for other structural methods.[23] Another interesting avenue in protein structural studies is laser-induced covalent labeling. In this technique, solvent-exposed sites of the protein are modified by hydroxyl radicals. Its combination with rapid mixing has been used in protein folding studies.[24]
Proteogenomics
In what is now commonly referred to as proteogenomics, peptides identified with mass spectrometry are used for improving gene annotations (for example, gene start sites) and protein annotations. Parallel analysis of the genome and the proteome facilitates discovery of post-translational modifications and proteolytic events,[25] especially when comparing multiple species.[26]
References
- Jürgen Cox and Matthias Mann (July 2011). "Quantitative, High-Resolution Proteomics for Data-Driven Systems Biology". Annual Review of Biochemistry. 80: 273–299. doi:10.1146/annurev-biochem-061308-093216. PMID 21548781.CS1 maint: uses authors parameter (link) – via Annual Reviews (subscription required)
- Nelson P. Barrera and Carol V. Robinson (July 2011). "Advances in the Mass Spectrometry of Membrane Proteins: From Individual Proteins to Intact Complexes". Annual Review of Biochemistry. 80: 247–71. doi:10.1146/annurev-biochem-062309-093307. PMID 21548785.CS1 maint: uses authors parameter (link) – via Annual Reviews (subscription required)
- Karas, M.; Bachmann, D.; Hillenkamp, F. (1985). "Influence of the Wavelength in High-Irradiance Ultraviolet Laser Desorption Mass Spectrometry of Organic Molecules". Analytical Chemistry. 57 (14): 2935–9. doi:10.1021/ac00291a042.
- Karas, M.; Bachmann, D.; Bahr, U.; Hillenkamp, F. (1987). "Matrix-Assisted Ultraviolet Laser Desorption of Non-Volatile Compounds". International Journal of Mass Spectrometry and Ion Processes. 78: 53–68. Bibcode:1987IJMSI..78...53K. doi:10.1016/0168-1176(87)87041-6.
- Dole M, Mack LL, Hines RL, Mobley RC, Ferguson LD, Alice MB (1968). "Molecular Beams of Macroions". Journal of Chemical Physics. 49 (5): 2240–2249. Bibcode:1968JChPh..49.2240D. doi:10.1063/1.1670391.
- Birendra N. Pramanik; A.K. Ganguly; Michael L. Gross (28 February 2002). Applied Electrospray Mass Spectrometry: Practical Spectroscopy Series. CRC Press. pp. 4–. ISBN 978-0-8247-4419-9.
- "Press Release: The Nobel Prize in Chemistry 2002". The Nobel Foundation. 2002-10-09. Retrieved 2011-04-02.
- Chait, Brian T. (2011). "Mass Spectrometry in the Postgenomic Era". Annu Rev Biochem. 80: 239–46. doi:10.1146/annurev-biochem-110810-095744. PMID 21675917. – via Annual Reviews (subscription required)
- Trauger, A. Sunia; W. Webb; G. Siuzdak (2002). "Peptide and protein analysis with mass spectrometry". Spectroscopy. 16 (1): 15–28. doi:10.1155/2002/320152.
- W. Wells, G. Wang & S. J. Baek (2006). "Comparative Study of Three Proteomic Quantitative Methods, DIGE, cICAT, and iTRAQ, Using 2D Gel- or LC−MALDI TOF/TOF". Journal of Proteome Research. 5 (3): 651–658. CiteSeerX 10.1.1.156.5802. doi:10.1021/pr050405o. PMID 16512681.
- Issaq, J. Haleem & T. D. Veenstra (2008). "Two-dimensional polyacrylamide gel electrophoresis (2D-PAGE): advances and perspectives". BioTechniques. 46 (5): 697–700. doi:10.2144/000112823. PMID 18474047.
- Bret, Cooper and J. Feng and W. Garrett (2010). "Relative, Label-free Protein Quantitation: Spectral Counting Error Statistics from Nine Replicate MudPIT Samples". Spectroscopy. 21 (9): 1534–1546. doi:10.1016/j.jasms.2010.05.001. PMID 20541435.
- Augustin, Scalbert and L. Brennan and O. Fiehn (2009). "Mass-spectrometry-based metabolomics: limitations and recommendations for future progress with particular focus on nutrition research". NCBI. 5 (4): 435–458. doi:10.1007/s11306-009-0168-0. PMC 2794347. PMID 20046865.
- P. Hernandez, M. Müller & R. D. Appel (2006). "Automated protein identification by tandem mass spectrometry: Issues and strategies". Mass Spectrometry Reviews. 25 (2): 235–254. Bibcode:2006MSRv...25..235H. doi:10.1002/mas.20068. PMID 16284939.
- Cravatt, Benjamin F.; Simon, Gabriel M.; Yates Iii, John R. (December 2007). "The biological impact of mass-spectrometry-based proteomics". Nature. 450 (7172): 991–1000. doi:10.1038/nature06525. ISSN 1476-4687.
- Slavov, Nikolai (2021-02-01). "Single-cell protein analysis by mass spectrometry". Current Opinion in Chemical Biology. 60: 1–9. doi:10.1016/j.cbpa.2020.04.018. ISSN 1367-5931.
- Slavov, Nikolai (2020-01-31). "Unpicking the proteome in single cells". Science. 367 (6477): 512–513. doi:10.1126/science.aaz6695. ISSN 0036-8075. PMID 32001644.
- Snijders AP, de Vos MG, Wright PC (2005). "Novel approach for peptide quantitation and sequencing based on 15N and 13C metabolic labeling". J. Proteome Res. 4 (2): 578–85. doi:10.1021/pr0497733. PMID 15822937.
- M. Miyagi & K. C. S. Rao (2007). "Proteolytic 18O-labeling strategies for quantitative proteomics". Mass Spectrometry Reviews. 26 (1): 121–136. Bibcode:2007MSRv...26..121M. doi:10.1002/mas.20116. PMID 17086517.
- Haqqani AS, Kelly JF, Stanimirovic DB (2008). Quantitative protein profiling by mass spectrometry using label-free proteomics. Methods Mol. Biol. Methods in Molecular Biology. 439. pp. 241–56. doi:10.1007/978-1-59745-188-8_17. ISBN 978-1-58829-871-3. PMID 18370108.
- Z. Zhang; D. L. Smith (1994). "Probing noncovalent structural features of proteins by mass spectrometry". Mass Spectrometry Reviews. 13 (5–6): 411–429. Bibcode:1994MSRv...13..411S. doi:10.1002/mas.1280130503.
- T. E. Wales & J. R. Engen (2006). "Hydrogen exchange mass spectrometry for the analysis of protein dynamics". Mass Spectrometry Reviews. 25 (1): 158–170. Bibcode:2006MSRv...25..158W. doi:10.1002/mas.20064. PMID 16208684.
- R.E. Iacob & J. R. Engen (2012). "Hydrogen Exchange Mass Spectrometry: Are We Out of the Quicksand?". Mass Spectrometry Reviews. 23 (6): 1003–1010. Bibcode:2012JASMS..23.1003I. doi:10.1007/s13361-012-0377-z. PMC 3389995. PMID 22476891.
- B. B. Stocks & L. Konermann (2009). "Structural Characterization of Short-Lived Protein Unfolding Intermediates by Laser-Induced Oxidative Labeling and Mass Spectrometry". Anal. Chem. 81 (1): 20–27. doi:10.1021/ac801888h. PMID 19055350.
- Gupta N.; Tanner S.; Jaitly N.; Adkins J.N.; Lipton M.; Edwards R.; Romine M.; Osterman A.; Bafna V.; Smith R.D.; et al. (2007). "Whole proteome analysis of post-translational modifications: Applications of mass-spectrometry for proteogenomic annotation". Genome Res. 17 (9): 1362–1377. doi:10.1101/gr.6427907. PMC 1950905. PMID 17690205.
- Gupta N.; Benhamida J.; Bhargava V.; Goodman D.; Kain E.; Kerman I.; Nguyen N.; Ollikainen N.; Rodriguez J.; Wang J.; et al. (2008). "Comparative proteogenomics: Combining mass spectrometry and comparative genomics to analyze multiple genomes". Genome Res. 18 (7): 1133–1142. doi:10.1101/gr.074344.107. PMC 2493402. PMID 18426904.
| Instrumentation |
|
|---|---|
| Techniques |
|
| Sampling |
|
| Calibration | |
| Prominent publications | |
| |