Michaelis–Arbuzov reaction
The Michaelis–Arbuzov reaction (also called the Arbuzov reaction) is the chemical reaction of a trivalent phosphorus ester with an alkyl halide to form a pentavalent phosphorus species and another alkyl halide. The picture below shows the most common types of substrates undergoing the Arbuzov reaction; phosphite esters (1) react to form phosphonates (2), phosphonites (3) react to form phosphinates (4) and phosphinites (5) react to form phosphine oxides (6).
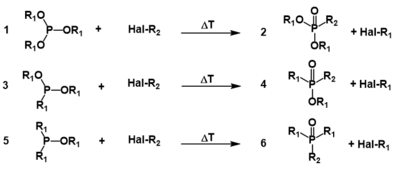
| Michaelis–Arbuzov reaction | |
|---|---|
| Named after | August Michaelis Aleksandr Arbuzov |
| Reaction type | Coupling reaction |
| Identifiers | |
| Organic Chemistry Portal | arbuzov-reaction |
| RSC ontology ID | RXNO:0000060 |
The reaction was discovered by August Michaelis in 1898,[1] and greatly explored by Aleksandr Arbuzov soon thereafter.[2][3] This reaction is widely used for the synthesis of various phosphonates, phosphinates, and phosphine oxides. Several reviews have been published.[4][5] The reaction also occurs for coordinated phosphite ligands, as illustrated by the demethylation of {(C5H5)Co[(CH3O)3P]3}2+ to give {(C5H5)Co[(CH3O)2PO]3}−, which is called the Klaui ligand.
Reaction mechanism
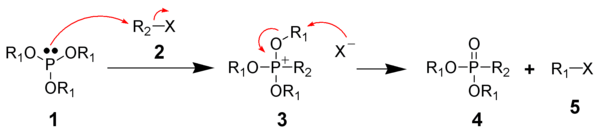
The Michaelis–Arbuzov reaction is initiated with the SN2 attack of the nucleophilic phosphorus species (1 - A phosphite) with the electrophilic alkyl halide (2) to give a phosphonium salt as an intermediate (3). These intermediates are occasionally stable enough to be isolated, such as for triaryl phosphites which do not react to form the phosphonate without thermal cleavage of the intermediate (200°C), or cleavage by alcohols or bases. The displaced halide anion then usually reacts via another SN2 reaction on one of the R1 carbons, displacing the oxygen atom to give the desired phosphonate (4) and another alkyl halide (5). This has been supported by the observation that chiral R1 groups experience inversion of configuration at the carbon center attacked by the halide anion. This is what is expected of an SN2 reaction.[6] Evidence also exists for a carbocation based mechanism of dealkylation similar to an SN1 reaction, where the R1 group initially dissociates from the phosphonium salt followed by attack of the anion.[5] Phosphite esters with tertiary alkyl halide groups can undergo the reaction, which would be unexpected if only an SN2 mechanism was operating. Further support for this SN1 type mechanism comes from the use of the Arbuzov reaction in the synthesis of neopentyl halides, a class of compounds that are notoriously unreactive towards SN2 reactions. Based on the principle of microscopic reversibility, the inert nature of the neopentyl halides towards the SN2 reaction indicates that an SN2 reaction is unlikely to be the mechanism for the synthesis of the neopentyl halides in this reaction. Substrates that cannot react through an SN2 pathway or an SN1 pathway generally do not react, which include vinyl and aryl groups. For example, the triaryl phosphites mentioned above generally do not react because they form stable phosphonium salts. Since aryl groups do not undergo SN1 and SN2 type mechanisms, triaryl phosphites lack a low energy pathway for decomposition of the phosphonium salt. An allylic rearrangement mechanism (SN2`) has also been implicated in allyl and propargyl halides.
Stereochemical experiments on cyclic phosphites have revealed the presence of both pentavalent phosphoranes and tetravalent phosphonium intermediates in chemical equilibrium being involved in the dealkylation step of the reaction using 31P NMR. The decomposition of these intermediates is driven primarily by the nucleophilicity of the anion. There exists many instances of the intermediate phosphonium salts being sufficiently stable that they can be isolated when the anion is weakly nucleophilic, such as with tetrafluoroborate or triflate anions.
Scope
Alkyl halide[5]
As a general guideline, the reactivity of the organic halide component can be listed as follows: (from most reactive to least reactive)

and

In general, tertiary alkyl haldies, aryl halides and vinyl halides do not react. There are notable exceptions to this trend, including 1,2-dichloroethene and trityl halides. Some activated aryl halides, often involving heterocycles have been known to undergo the reaction. Iodobenzene and substituted derivatives have been known to undergo the reaction under photolytic conditions. Secondary alkyl halides often do not react well, producing alkenes as side-products. Allyl and propargyl halides are also reactive, but can proceed through an SN2 or an SN2` mechanism. Reaction with primary alkyl halides and acyl halides generally proceed smoothly. Carbon tetrachloride interestingly enough, only undergoes the reaction a single time with chloroform being inert to the reaction conditions. When a halide atom is found in the ester chain off of the phosphorus atom, isomerization to the corresponding Arbuzov product has been known without addition of an alkyl halide.
The Perkow reaction is a competing reaction pathway for α-bromo- and α-chloroketones. Under the reaction conditions a mixture of the Perkow product and the normal Arbuzov product occur, usually favoring the Perkow product by a significant amount. Using higher temperatures during the reaction can lead to favoring of the Arbuzov product. The reaction of α-Iodoketones give only the Arbuzov product.[7] Other methods of producing β-ketophosphonates have been developed.[8]
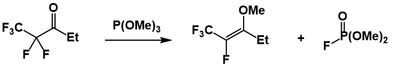
The reaction of trivalent phosphorus compounds with alkyl fluorides is abnormal. One example of this reactivity is shown below.
Phosphorus reactant[5]
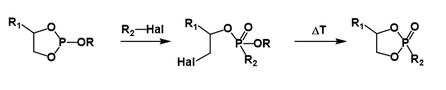
The general form of the trivalent phosphorus reagent can be considered as follows: with A and B generally being alkyl, alkoxy or aryloxy groups. Electron-withdrawing groups are known to slow down the rate of the reaction, with electron donating groups increasing the rate of the reaction. This is consistent with initial attack of the phosphorus reagent on the alkyl halide as the rate-determining step of the reaction. The reaction proceeds smoothly when the R group is aliphatic. When all of A, B and R are aryl groups, a stable phosphonium salt is formed and the reaction proceeds no further under normal conditions. Heating to higher temperatures in the presence of alcohols has been known to give the isomerization product. Cyclic phosphites generally react to eject the non-cyclic OR group, though for some 5-member rings additional heating is required to afford the final cyclic product.

Phosphite salts (Ex: R = Na) can also undergo the reaction with precipitation of the corresponding Na-halide salt. Amidophosphites and silyloxyphosphites have been used before to yield amidophosphonates and phosphinic acids.

An Arbuzov type rearrangement can also occur where the O from an OR group acts as the leaving group in the initial SN2 attack of the phosphorus. This is only known to occur when A and B are Cl.
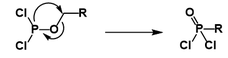
Phosphite esters are the least reactive class of reagents used in this reaction. They react to produce phosphonates. They require the most heating for the reaction to occur (120°C - 160°C is common). This high temperature allows for fractional distillation to be employed in the removal of the alkyl halide produced, though excess of the starting alkyl halide can also be used. Solvents are often not used for this reaction, though there is precedent for the improvement of selectivity with its usage.
Phosphonites are generally more reactive than phosphite esters. They react to produce phosphinates. Heating is also required for the reaction, but pyrolysis of the ester to an acid is a common side reaction. The poor availability of substituted phosphonites limits the usage of this class of reagent in the Arbuzov reaction. Hydroxy, thiol, carboxylic acid, primary and secondary amine functional groups cannot be used with phosphonites in the reaction as they all react with the phosphonite.
Phosphinites are the most reactive class of reagents used in this reaction. They react to produce phosphine oxides. They often require very little heating (45°C) for the reaction to occur and have been known to self-isomerize without the presence of alkyl halides.
References
- Michaelis, A.; Kaehne, R. (1898). "Ueber das Verhalten der Jodalkyle gegen die sogen. Phosphorigsäureester oder O-Phosphine". Berichte. 31: 1048–1055. doi:10.1002/cber.189803101190.
- Arbuzov, A. E. (1906). J. Russ. Phys. Chem. Soc. 38: 687. Missing or empty
|title=(help) - Arbuzov, A. E. (1906). Chem. Zentr. II: 1639. Missing or empty
|title=(help) - Arbuzov, B. A. (1964). "Michaelis–Arbusow- und Perkow-Reaktionen". Pure Appl. Chem. 9 (2): 307–353. doi:10.1351/pac196409020307.
- Bhattacharya, A. K.; Thyagarajan, G. (1981). "Michaelis–Arbuzov rearrangement". Chem. Rev. 81 (4): 415–430. doi:10.1021/cr00044a004.
- Gerrard, W.; Green, W. J. (1951). "568. Mechanism of the formation of dialkyl alkylphosphonates". J. Chem. Soc.: 2550. doi:10.1039/jr9510002550.
- Jacobsen, H. I.; Griffin, M. J.; Preis, S.; Jensen, E. V. (1957). "Phosphonic Acids. IV. Preparation and Reactions of β-Ketophosphonate and Enol Phosphate Esters". J. Am. Chem. Soc. 79 (10): 2608. doi:10.1021/ja01567a067.
- Nagata, W.; Wakabayashi, T.; Hayase, Y. (1988). "Diethyl 2-(cyclohexylamino)vinylphosphonate". Organic Syntheses.CS1 maint: multiple names: authors list (link); Collective Volume, 6, p. 448
External links
- Ford-Moore, A. H.; Perry, B. J. Organic Syntheses, Coll. Vol. 4, p. 325 (1963); Vol. 31, p. 33 (1951). (Article)
- Davidsen, S. K.; Phllips, G. W.; Martin, S. F. Organic Syntheses, Coll. Vol. 8, p. 451 (1993); Vol. 65, p. 119 (1987). (Article)
- Enders, D.; von Berg, S.; Jandeleit, B. Organic Syntheses, Coll. Vol. 10, p. 289 (2004); Vol. 78, p. 169 (2002). (Article)