Metalloprotease inhibitor
Metalloprotease inhibitors are cellular inhibitors of the Matrix metalloproteinases (MMPs).[1] MMPs belong to a family of zinc-dependent neutral endopeptidases.[2] These enzymes have the ability to break down connective tissue. The expression of MMPs is increased in various pathological conditions like inflammatory conditions, metabolic bone disease, to cancer invasion, metastasis and angiogenesis. Examples of diseases are periodontitis, hepatitis, glomerulonephritis, atherosclerosis, emphysema, asthma, autoimmune disorders of skin and dermal photoaging, rheumatoid arthritis, osteoarthritis, multiple sclerosis, Alzheimer's disease, chronic ulcerations, uterine involution, corneal epithelial defects, bone resorption and tumor progression and metastasis.[2][3] Due to the role of MMPs in pathological conditions, inhibitors of MMPs may have therapeutic potential.[3] Several other proteins have similar inhibitory effects, however none as effective (netrins, procollagen C-terminal proteinase enhancer (PCPE), reversion-inducing cysteine-rich protein with Kazal motifs (RECK) and tissue factor pathway inhibitor (TFPI-2)). They might have other biological activities which have yet been fully characterised.[4]
MMP inhibitors can broadly be subdivided into non-synthetic (e.g. endogenous) or synthetic.[2] Several potent MMP inhibitors have been identified, including hydroxymates, thiols, carbamoylphosphonates, hydroxyureas, hydrazines, β-lactams, squaric acids and nitrogenous ligands.[5]
There are three classes of commonly used inhibitors for metalloproteinases.
- In vitro, EDTA, 1,10-phenanthroline and other chelating compounds lower the concentration of metal to the point where the metal is removed from the enzyme active site.
- Classical lock and key inhibitors such as phosphoramidon and bestatin bind tightly by approximating the transition state of the hydrolysis of the peptide, preventing it from acting on other substrates.
- Protein inhibitors such as α2-macroglobulin are known to work with metalloproteinases.
History
The first generation of MMP inhibitors were based on the structure of the collagen molecule. This group of inhibitors contain a hydroxamate (-CONHOH) group that binds the zinc atom in the active site of the MMP enzyme.[6] The first MMP inhibitors that were tested in patients were Ilomastat and Batimastat, hydroxamate-based MMP inhibitors.[6][7] However, neither compound showed good oral bioavailability.[6]
Thus far, Periostat (active ingredient is doxycycline hyclate) is the only MMP inhibitor that has been approved by the U.S. Food and Drug Administration (FDA). It is used for the treatment of periodontitis. Other MMP inhibitors have exhibited serious side effects during preclinical trials. These side effects are caused by insufficient selectivity. Most MMP inhibitors are unable to target specific MMPs connected to specific pathological conditions. Instead, they inhibit multiple MMPs, some of which have protective functions or are not related to pathology.[5]
MMPs have been regarded as promising targets for cancer therapy. Preclinical studies investigating the efficacy of MMP suppression in tumor models were encouraging. Following these results, clinical studies were conducted but turned out to be disappointing. Recent studies have shown that MMPs may even have paradoxical roles in tumor progression. MMPs seem to have tumor-promoting effects as well as tumor suppressive effects dependent on different contexts.[8]
Mechanism of action
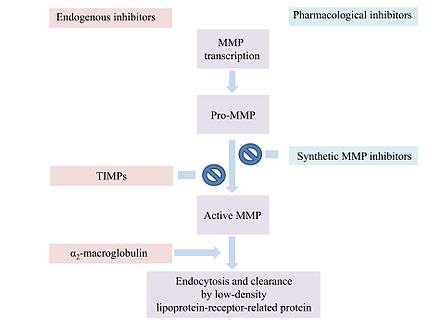
Most MMP inhibitors are chelating agents. The inhibitor binds to the zinc at the active center of the enzyme, thereby blocking its activity. Other inhibitor mechanisms are possible.[1]
α2-Macroglobulin (α2M) is a protease inhibitor which inhibits activated MMPs. α2M and MMP form a complex which is able to inactivate the MMP.[9]
MMPs are associated with the cell surface or bound to the extracellular matrix which prevents them from diffusing away and keeps the MMP under control of the cell. One mechanism to inhibit MMP activity is by dislodging the enzymes from their receptors. Gold salts bind to a heavy metal site distinct form the zinc-containing active center, which inhibits their activity. MMP activity can be decreased by binding to the cleavage site on the substrate e.g. catechin.[1]
Two molecular features of most MMP inhibitors are responsible for the affinity. One is a chelating moiety that interacts with the zinc ion and the other is a hydrophobic extension from the catalytic site that project into S1’ pocket (P1’ group) of the metalloproteinase. The structural difference MMPs’ is mainly in the S1’ side and by modifying the P1’ group, inhibitor selectivity can be developed.[10]
Drug development
Various potential MMP inhibitors will be explained in the following sections, including information on their development, structure-activity relationship and pharmacokinetics.
Pioneering hydroxamate structures
The first generation of MMP inhibitors were based on the structure of the collagen molecule. In the design of these inhibitors, the basic protein backbone of collagen is maintained but the amide bond is replaced with a zinc-binding group.[11] This group of inhibitors contain a hydroxamate (-CONHOH) group that binds the zinc atom in the active site of the MMP enzyme, therefore this group is called "hydroxamate-based MMP inhibitors“.[6] An example can be seen in Marimastat, a first generation inhibitor, which has a similar backbone and sidechain format to collagen.
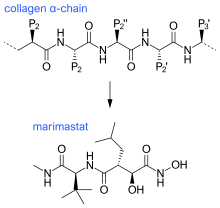
Ilomastat and batimastat were the first two MMP inhibitors to be tested in patients. These are both hydroxamate-based MMP inhibitors and have similar overall structures.
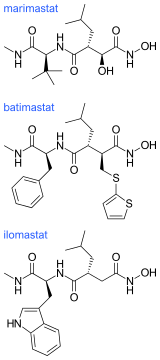
The hydroxamate-based MMP inhibitors display an excellent anticancer activity in tumor cells but the clinical performances of these compounds were disappointing. A factor contributing to this disappointment was that they are broad-spectrum inhibitors of many MMP sub-types that can in many cases also inhibit members of the ADAMs protease family. When they were tested in patients they induced dose-limiting muscular and skeletal pain in a number of the patients.[5][7] Only when the structures of the MMP inhibitors could be adjusted to impart selectivity and abolish toxicity, would they achieve clinical impact in cancer chemotherapy.[7]
New generation hydroxamate-based inhibitors
The pioneering hydroxamate-based inhibitors were followed by a set of 'new generation' molecules with features including a substituted aryl, a sulfonamide and a hydroxamate zinc-binding group.
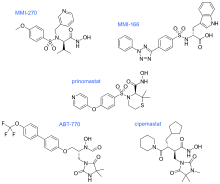
In MMI-270 there is also an amino acid sidechain-type substituent on the carbon that is α to the hydroxamate, along with a sidechain on the sulfonamide (which was later shown to be unnecessary). The N-arylsulfonyl-α-aminoacid hydroxamate of MMI-270 mimics the marimastat succinate motif. Cipemastat, which was developed as an MMP-1, -3 and -9 collagenase inhibitor for the treatment of rheumatoid- and osteo-arthritis, also has the marimastat succinate motif. Its clinical trial was terminated prematurely.
MMI-166 has an N-arylsulfonyl-α-aminocarboxylate zinc-binding group, different from the hydroxamate-zinc binding group seen in MMI-270 and Cipemastat. It also has a triaryl substitution that the other structures didn‘t have. ABT-770 and Prinomastat also have an aryl substitution. In ABT-770 the two phenyl rings are directly connected but in Prinomastat the two phenyl rings are connected by an oxygen atom, forming a diphenylether. These three permutations direct the SAR away from MMP-1 and toward the „deep pocket“ MMPs such as the gelatinases. ABT-770 shows anticancer activity in animal models, but it is easily metabolised to an amine metabolite that causes phospholipidosis. MMI-166 has shown anticancer activity in numerous animal models, but there is no data available of its clinical performance. Prinomastat on the other hand, is one of the best studied MMP inhibitors. It showed excellent preclinical animal anticancer efficacy, but a recurring limitation to these hydroxamates (Prinomastat in particular) is drug metabolism including loss of the hydroxamate zinc-binding group.
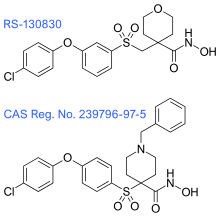
These inhibitors were followed by the next group of hydroxamate-based inhibitors, which focus on the suppression of metabolism, minimization of MMP-1 inhibitory activity and the control of subtype selectivity, by structure-based design. The tetrahydropyran in RS-130830 introduces a steric block that suppresses metabolism, which would fix the problem that the previous generation of inhibitors showed. The outcome of its clinical evaluation has not yet been disclosed. 239796-97-5 has improved ADME and MMP-1 selectivity properties and has shown excellent oral efficacy in an animal model of osteoarthritis. Although, the therapeutic objectives for these inhibitors is not cancer, like it has been for most of the MMP inhibitors.[7]
New generation thiol-based inhibitors
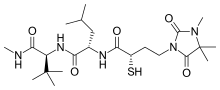
Rebimastat is a broad spectrum MMP inhibitor with a thiol zinc-binding group. It has oral bioavailability and is a collagen non-peptide mimetic. Rebimastat has some selectivity as it doesn't inhibit all the MMPs operations. The metalloproteinases that release TNF-alpha, TNF-II, L-selectin, IL-1-RII and IL-6 are for example not inhibited by Rebimastat.
In phase I of clinical trials, there was no sign of dose-dependent joint toxicity and a disease stabilization. Arthralgia was noted in phase II early breast cancer trials, which was connected to MMP inhibitor toxicity. Rebimastat was used in a Paclitaxel/Carboplatin treatment in phase III. The results of the trial was a higher incidence of adverse reactions, without survival benefit.

Clinical trials for Tanomastat, an alfa-((phenylthio)methyl)carboxylate, showed similar results. It showed good disease stability and tolerance in Phase I solid tumor trials and good tolerance in advanced cancer in combination with Etoposide. However, its efficacy was not proven to be adequate. Tanomastat showed significant hepatotoxicity in a cancer therapy combined with Cisplatin and Etoposide, although in a treatment with Doxorubicin it showed good tolerance and lowered toxicity with 5-fluorouracil and Leucovorin.
Many compounds in the thiol zinc-binding groups have good water solubility and are air stable in plasma and these groups will be continued in MMP inhibitor designing.[7]
Pyrimidine-based inhibitors
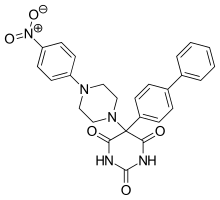
Ro 28-2653 is highly selective for MMP-2, MMP-9 and membrane type 1 (MT-1)-MMP. It is an antitumor and antiangiogenic agent with oral bioavailability. Inhibition of TACE and MMP-1 are linked to the musculoskeletal side effects seen in hydroxamate metalloproteinase inhibitors, but this compound spares the enzymes.[12] It has been shown to diminish tumor growth in nasal cancer in rats as well as prostate cancer cell cultures. The compound only has moderate effect on mice's adipose tissue and no alteration on joints. Based on this, it is concluded that class of inhibitors is less likely to trigger neuromuscular adverse effects. On the active site of the structure is a pyrimidinetrione chelation and the phenyl and piperidynil section occupy the S1’ and S2’ binding pockets of MMP-8.
Compound 556052-30-3 is similar to Ro 28-2653 but incorporates a 4-((2-methylquinolin-4-yl)methoxy)phenyl sidechain that is TACE selective.
5-(spiropyrrolidin-5-yl)pyrimidinetrione is a compound named 848773-43-3 that is a potent MMP-2, MMP-9 and MMP-13 inhibitor that spares MMP-1 and TACE.
By substituting 1,3,4-oxadiazol-2-yl heteroaryl at C-4’ of the diphenylether segment to accomplish MMP-13 selectivity over MT-1 MMP, made the compound 420121-84-2. The compound has IC50 (half maximal inhibitory concentration) of 1 nM for MMP-13.
I125-radioable pyrimidinetriones that have similar structure have been made to be used in MMP-9 elevated atherosclerosis and elevated MMP-2 and MMP-9 cancers.
This class of MMP inhibitors is easy to synthesize and are potent enough for clinical valuation.
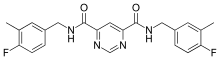

Compound 544678-85 is the latest pyrimidine based inhibitor, the compound is a pyrimidine-4,6-dicarboxamide that is very potent and MMP-13 selective. The compound has a specificity loop that within the S1’ pocket and its 3-methyl-4-fluoro group is proximal enough to the zinc to change the water entity. These compounds have good oral bioavailability and properties that promote them to be a good candidate for a subtype inhibitor of MMP-13 based diseases and future development.[7]
Pyrimidine dicarboxamides are highly selective MMP-13 inhibitors. In the S1’ pocket of MMP-13 is an S1’ side pocket that is unique to the matrix metalloproteiase. Pyrimidine dicarboxamides bind to this side pocket, which increases the selectivity. The role of MMP-13 is cleaving fibrillar collagen at neutral pH and higher mRNA levels of MMP-13 is detected in breast carcinoma and osteoarthritis joints.
The pyrimidine dicarboxamide inhibitor example in the image does not interact with the catalytic zinc ion but rather binds to the S1’ side pocket.[10] One pyridyl arm is situated to the entrance of the S1’ pocket while the other pyridyl arm goes through the S1’ pocket into the side pocket.[13]
Hydroxypyrone-based inhibitors
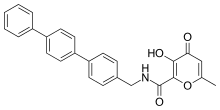
Potent and selective MMP-3 inhibitors have been developed by using a hydroxypyrone as the zinc binding group. By attaching an aryl backbone to the 2-position of the pyrone ring, more selectivity was gained. On the hydroxypyrone ring, three positions are available to attach backbones, position 2-, 5- and 6-.[14]
Hydroxypyrone-based MMP inhibitors are structurally corresponding to the pyrimidinetriones. A recent inhibitor is the compound 3-hydroxypyran-4-one nominated 868368-30-3. It is MMP-3 selective and its 0,0-bidentate chelation of zinc is the structural part proposed to be responsible for the MMP recognition.[7]
Phosphorus-based inhibitors
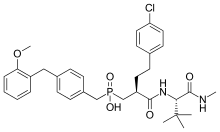
Investigation on MMP inhibitors with phosphorus based zinc binding groups focused on α-biphenylsulfonylamino phosphonates. These inhibitors bind through two phosphonate oxygen atoms. Phosphonate inhibitors have been developed that exhibit selectivity for MMP-8 over other MMPs. Selective MMP-8 inhibitors could be useful in the treatment of acute liver disease and multiple sclerosis[15] Phosphinic MMP inhibitors have been reported to target MMP-11 and MMP-13. MMP-13 plays a role in cartilage degradation in osteoarthritis. These phosphinate MMP inhibitors contain phenyl segments that are thought to be responsible for the selectivity to MMP-13. The phosphinic group of those inhibitors (R1R2 (O)OH) binds as a zinc ligand. R1 and R2 substituents affect the inhibition potency.[7]
Phosphinate inhibitors have been developed that showed high selectivity for MMP-11. Derivatives based on phenyl rings showed the best selectivity. MMP-11 could be a useful target for tumorgenesis in breast cancer.
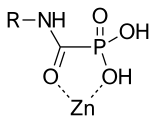
Phosphorus-based inhibitors with carbamoyl phosphonate zinc binding groups do not bind through two oxygen of the phosphonate. Carbamoyl phosphonate zinc binding groups binds Zn2+ through the oxygen of the phosphonate and the oxygen of the alpha carbonyl to the phosphonate. This binding forms a 5-members chelate ring that looks similar to the binding of hydroxamic acid.
The amide bond of the carbamoyl phosphonate provide a hydrogen bond donor for protein interactions and the amide group has an electron donating ability that provides strong Zn2+ chelation.
The carbamoyl phosphonate zinc binding groups have a net negative charge that hinders cell penetration of these inhibitors and restricts them to the extracellular space. This cell penetration prevention contributes to the low toxicity of these inhibitors. Inhibitors with a carbamoyl phosphonate zinc-binding group are selective for MMP-2. MMP-2 could be a useful target for tumor invasion and angiogenesis. A carbamoyl phosphonate inhibitor has been developed that affects MMP-2 and MMP-9 sparing other MMPs. This compound showed inhibitory activity on cell invasion and tumor colonization. In in vivo studies, this inhibitor showed efficacy with oral dosing and administration into the abdominal cavity (intraperitoneal). It shows slow absorption, rapid elimination and low oral bioavailability. Prolonged absorption contributes to sustained efficacy. Inhibitors with carbamoyl phosphonate zinc binding groups are water-soluble at physiological pH.[15]
Tetracycline-based inhibitors
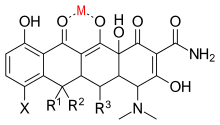
Tetracyclines are antibiotics that also exhibit MMP inhibitory activity. They chelate Zn2+ ion, thereby inhibiting MMP activity. It is believed that tetracyclines also effect MMP expression and proteolytic activity.[16]
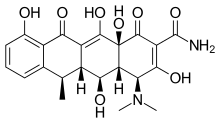
Doxycycline is a semi-synthetic tetracycline that has been studied for dental and medical applications. Its effects on diseases like periodontitis and cancer has been investigated.[2] Doxycycline is nearly completely absorbed with a bioavailability about 95% in average and a 20% reduction with co-administration of food. Its volume of distribution is 50–80 L (0,7 L/kg). Protein binding is 82–93 %. It is excreted in urine and in feces. Doxycycline is available in oral and intravenous form.[17] Doxycycline exhibited inhibitory activity on MMP-2 and MMP-9.[2] The expression and activity of MMP-2 and MMP-9 are often elevated in human cancer. The increased expression and activity correlates with advanced tumor stage, increased metastasis and prognosis.[8]
Chemically modified tetracyclines (CMT) have been developed to explore their inhibitory potential. Most studies in tetracyclines and CMTs showed that they can inhibit MMP activity.
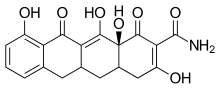
One CMT called COL-3 or metastat has been demonstrated to be a potent MMP inhibitor. COL-3 features a tetracycline scaffold that is unsubstituted on positions C4–C9.[16]
Advantages of CMT over conventional tetracyclines are that chronic use does not result in gastrointestinal toxicity and higher plasma levels can be achieved for extended time span reducing administration frequency.
The pharmacokinetics of COL-3 has been studied in rats. COL-3 is absorbed slowly from the gastrointestinal tract. 3% are excreted through the urinary tract while 55–66% is excreted in feces. The drug is highly lipophilic and able to cross the blood brain barrier at higher doses. COL-3 accumulates in higher concentration in heart tissue and testis.
In clinical trials plasma protein binding has been shown to be high (~94,5%). Most COL-3 binds to serum albumin.[2]
Endogenous inhibitors
MMP activity is regulated at various levels for example by endogenous inhibitors like α2-macroglobulin and the tissue inhibitors of metalloproteinases (TIMPs).[18]
α2-macroglobulin regulates a broad spectrum of proteases, while TIMPs are more specific endogenous MMP inhibitors.
α2-macroglobulin is an abundant plasma protein that acts in tissue fluids.[8] The plasma glycoprotein consists of four subunits.[19] α2-macroglobulin does not inhibit the activation of MMPs or the MMPs themselves.[8] It entraps proteinases like MMPs and forms a complex with them.[19] The complex is endocytosed and cleared by a low-density lipoprotein-receptor-related protein.[8]
In humans, 4 different TIMPs have been found. They are secreted proteins of low-molecular weight. TIMPs bind noncovalently to the active site of MMPs.[18] Changes of TIMP levels are considered to play a role in pathological conditions associated with unbalanced MMP activities.[19] TIMPs consist of 184-194 amino acids. These inhibitors are subdivided into two domains N-terminal and C-terminal.[19] The N-terminal regions of the four TIMPs share a common structure. They all contain twelve cysteine residues that form six disulfide bonds. These bonds are critical for the conformation of the N-terminal and its MMP-inhibitory activities. The C-terminals of the TIMPs differ from each other.[8] The N-terminal subunit is capable of inhibiting MMPs. The TIMP molecule form fits into the active site of an MMP. The TIMP contact the catalytic cleft of the MMP in a similarly as a substrate. TIMPs inhibit all MMPs except TIMP-1 which does not inhibit MT-1-MMP.[19]
There are some differences in the inhibitory preferences of TIMPs. TIMP-1 for example favors to inhibit MMP-9. Other examples are TIMP-2 and TIMP-4 which are more potent MMP-2 inhibitors than MMP-9 inhibitors.[8]
TIMPs could potentially be useful against illnesses like cardiovascular disease and cancer. The application of TIMPs as therapeutic instrument through gene therapy or direct protein application is still in early stages of development.
It is preferable to inhibit specific MPPs that play a role in pathological conditions. Since TIMPs inhibit multiple MMPs it is desirable to develop engineered TIMPs with altered specificity.[19]
Current status
The primary goal of MMP inhibitor design is selectivity. The targeting of specific MMPs is expected to improve efficacy and prevent side effects like musculoskeletal syndrome (MSS).[15] 3D structures of MMP inhibitors provide a source of insight of the structural relationships for selectivity. High throughput screening can as well increase the chances of discovering inhibitors with high selectivity.[13]
See also
- Matrix metalloproteinase
- MMP inhibitors
- Collagen
- Cancer
- Periodontitis
- Arthritis
- Rheumatoid arthritis
- Osteoarthritis
- Tetracycline
- Periostat
References
- Frederick, W. (1999). "Matrix Metalloproteinase Inhibition: From The Jurassic To The Third Millennium". Ann N Y Acad Sci. 878 (1): 388–403. Bibcode:1999NYASA.878..388W. doi:10.1111/j.1749-6632.1999.tb07697.x. PMID 10415743.
- Acharya, M. R.; Venitz, J.; Figg, W. D.; Sparreboom, A. (2004). "Chemically modified tetracyclines as inhibitors of matrix metalloproteinases". Drug Resistance Updates. 7 (3): 195–208. doi:10.1016/j.drup.2004.04.002. PMID 15296861.
- Whittaker, Mark; Ayscough, Andrew (2001). "Matrix metalloproteinases and their inhibitors- current status and future challenges". Celltransmissions. 17 (1): 3–14.
- Baker, Andrew; Dylan R. Edwards; Gillian Murphy (October 2002). "Metalloproteinase inhibitors: biological actions and therapeutic opportunities". J Cell Sci. 115 (19): 3719–3727. doi:10.1242/jcs.00063. PMID 12235282.
- Durrant, J. D.; de Oliveira, C. A. F.; McCammon, J. A. (2011). "Pyrone-Based Inhibitors of Metalloproteinase Types 2 and 3 May Work as Conformation-Selective Inhibitors". Chemical Biology & Drug Design. 78 (2): 191–198. doi:10.1111/j.1747-0285.2011.01148.x. PMC 3135671. PMID 21609408.
- Brown, P. D. (1997). "Matrix metalloproteinase inhibitors in the treatment of cancer". Medical Oncology. 14 (1): 1–10. doi:10.1007/BF02990939. PMID 9232605.
- Fisher, J. F.; Mobashery, S. (2006). "Recent advances in MMP inhibitor design". Cancer Metastasis Rev. 25 (1): 115–136. doi:10.1007/s10555-006-7894-9. PMID 16680577.
- Hua, H.; Li, M.; Luo, T; Yin, Y.; Jiang, Y (2011). "Matrix Metalloproteinases in Tumorigenesis: an Evolving Paradigm". Cellular and Molecular Life Sciences. 68 (23): 3853–3868. doi:10.1007/s00018-011-0763-x. PMID 21744247.
- Chen, J.; Chu, Y.; Cao, J.; Wang, W.; Liu, J.; Wang, J. (2011). "Effects of T-2 toxin and selenium on chondrocyte expression of matrix metalloproteinases (MMP-1, MMP-13), α2-macroglobulin (α2M) and TIMPs". Toxicology in Vitro. 25 (2): 492–499. doi:10.1016/j.tiv.2010.12.001. PMID 21144892.
- Engel, C. K.; Pirard, B.; Schimanski, S.; Kirsch, R.; Habermann, J.; Klingler, O.; Wendt, K. U. (2005). "Structural Basis for the Highly Selective Inhibition of MMP-13". Chemistry & Biology. 12 (2): 181–189. doi:10.1016/j.chembiol.2004.11.014. PMID 15734645.
- Whittaker, M.; Floyd, C. D.; Brown, P.; Gearing, A. J. H. (1999). "Design and therapeutic application of matrix metalloproteinase inhibitors". Chem. Rev. 99 (9): 2735–2776. doi:10.1021/cr9804543. PMID 11749499.
- Maquoi, E.; Sounni, N. E.; Devy, L.; Olivier, F.; Frankenne, F.; Krell, H. W.; Noel, A. (2004). "Anti-invasive, antitumoral, and antiangiogenic efficacy of a pyrimidine-2,4,6-trione derivative, an orally active and selective matrix metalloproteinases inhibitor". Clin Cancer Res. 10 (12): 4038–4047. doi:10.1158/1078-0432.ccr-04-0125. PMID 15217936.
- Pirard, B (2007). "Insight into the structural determinants for selective inhibition of matrix metalloproteinases". Drug Discovery Today. 12 (15–16): 640–646. doi:10.1016/j.drudis.2007.06.003. PMID 17706545.
- Yan, Y.-L.; Miller, M. T.; Cao, Y.; Cohen, S. M. (2009). "Synthesis of hydroxypyrone- and hydroxythiopyrone-based matrix metalloproteinase inhibitors: Developing a structure–activity relationship". Bioorganic & Medicinal Chemistry Letters. 19 (7): 1970–1976. doi:10.1016/j.bmcl.2009.02.044. PMC 2833267. PMID 19261472.
- Jacobsen, J. A.; Major Jourden, J. L.; Miller, M. T.; Cohen, S. M. (2010). "To bind zinc or not to bind zinc: An examination of innovative approaches to improved metalloproteinase inhibition". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1803 (1): 72–94. doi:10.1016/j.bbamcr.2009.08.006. PMID 19712708.
- Zakeri, B.; Wright, G. D. (2008). "Chemical biology of tetracycline antibiotics". Biochemistry and Cell Biology. 86 (2): 124–136. doi:10.1139/o08-002. PMID 18443626.
- Agwuh, K. N.; MacGowan, A. (2006). "Pharmacokinetics and pharmacodynamics of the tetracyclines including glycylcyclines". Journal of Antimicrobial Chemotherapy. 58 (2): 256–265. doi:10.1093/jac/dkl224. PMID 16816396.
- Rundhaug, J. E. (2003). "Matrix Metalloproteinases, Angiogenesis, and Cancer". Clin Cancer Res. 9 (2): 551–554. PMID 12576417.
- Nagase, H.; Visse, R.; Murphy, G. (2006). "Structure and Function of Matrix Metalloproteinases and TIMPs". Cardiovascular Research. 69 (3): 562–573. doi:10.1016/j.cardiores.2005.12.002. PMID 16405877.