LIG1
DNA ligase 1 is an enzyme that in humans is encoded by the LIG1 gene. DNA ligase I is the only known eukaryotic DNA ligase involved in both DNA replication and repair, making it the most studied of the ligases.
Discovery
It was known that DNA replication occurred through the breakage of the double DNA strand, but the enzyme responsible for ligating the strands back together, and mechanism of action, was unknown until Lehman, Gellert, Richardson, and Hurwitz laboratories, made significant contributions to the discovery of DNA ligase in 1967.[5]
Recruitment and regulation
The LIG1 gene encodes a, 120kDa enzyme, 919 residues long, known as DNA ligase I. The DNA ligase I polypeptide contains an N-terminal replication factory-targeting sequence (RFTS), followed by a nuclear localization sequence (NLS), and three functional domains.[6] The three domains consist of an N-terminal DNA binding domain (DBD), and catalytic nucleotidyltransferase (NTase), and C-terminal oligonucleotide / oligosaccharide binding (OB) domains. Although the N-terminus of the peptide has no catalytic activity it is needed for activity within the cells. The N-terminus of the protein contains a replication factory-targeting sequence that is used to recruit it to sites of DNA replication known as replication factories.
Activation and recruitment of DNA ligase I seem to be associated with posttranslational modifications. N-terminal domain is completed through phosphorylation of four serine residues on this domain, Ser51, Ser76, and Ser91 by cyclin-dependent kinase (CDK) and Ser66 by casein kinase II (CKII). Phosphorylation of these residues (Ser66 in particular) has been shown to possibly regulate the interaction between the RFTS to the proliferating cell nuclear antigen (PCNA) when ligase I is recruited to the replication factories during S-phase.[6][7] Rossi et al. proposed that when Ser66 is dephosphorylated, the RFTS of ligase I interact with PCNA, which was confirmed in vitro by Tom et al. Both data sets provide plausible evidence the N-terminal region of ligase I plays a regulatory role in the enzymes in vivo function in the nucleus.[7][8] Moreover, the identification of a cyclin binding (Cy) motif in the catalytic C-terminus domain was shown by mutational analysis to play a role in the phosphorylation of serines 91 and 76. Together, the N-terminal serines are substrates of the CDK and CKII, which appear to play an important regulatory role DNA ligase I recruitment to the replication factory during S-phase of the cell cycle.[6][9]

Function and mechanism
LIG1 encodes DNA ligase I, which functions in DNA replication and the base excision repair process.[10]
Eukaryotic DNA ligase 1 catalyzes a reaction that is chemically universal to all ligases. DNA ligase 1 utilizes adenosine triphosphate (ATP) to catalyze the energetically favorable ligation events in both DNA replication and repair. During the synthesis phase (S-phase) of the eukaryotic cell cycle, DNA replication occurs. DNA ligase 1 is responsible for joining Okazaki fragments formed during discontinuous DNA synthesis on the DNA's lagging strand after DNA polymerase δ has replaced the RNA primer nucleotides with DNA nucleotides. If the Okazaki fragments are not properly ligated together, the unligated DNA (containing a ‘nick’) could easily degrade to a double strand break, a phenomenon known to cause genetic mutations. In order to ligate these fragments together, the ligase progresses through three steps:
- Addition of an adenosine monophosphate (AMP) group to the enzyme, referred to as adenylylation,
- Adenosine monophosphate transfer to the DNA and
- Nick sealing, or phosphodiester bond formation.[8][11]
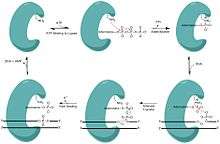
During adenylylation, there is a nucleophilic attack on the alpha phosphate of ATP from a catalytic lysine resulting in the production of inorganic pyrophosphate (PPi) and a covalently bound lysine-AMP intermediate in the active site of DNA ligase 1.
During the AMP transfer step, the DNA ligase becomes associated with the DNA, locates a nick and catalyzes a reaction at the 5’ phosphate site of the DNA nick. An anionic oxygen on the 5’ phosphate of the DNA nick serves as the nucleophile, attacking the alpha phosphate of the covalently bound AMP causing the AMP to be covalently bound intermediate (DNA-AMP intermediate).
In order for the phosphodiester bond to be formed, the DNA-AMP intermediate must be cleaved off. To accomplish this task, there is a nucleophilic attack on the 5’-phosphate from the upstream 3’-hydroxyl which results in the formation of the phosphodiester bond. During this nucleophilic attack, the AMP group is pushed off the 5’ phosphate as the leaving group allowing for the nick to seal and the AMP to be released, completing one cycle of DNA ligation.
Under suboptimal conditions the ligase can disassociate from the DNA before the full reaction is complete. It has been shown that magnesium levels can slow the nick sealing process, causing the ligase to disassociate from the DNA, leaving an aborted adenylylated intermediate incapable of being fixed without the aid of a phosphodiesterase. Aprataxin (a phosphodiesterase) has been shown to act on aborted DNA intermediates via hydrolysis of the AMP-phosphate bond, restoring the DNA to its initial state before the ligase had reacted.[12][13]
Role in damaged base repair
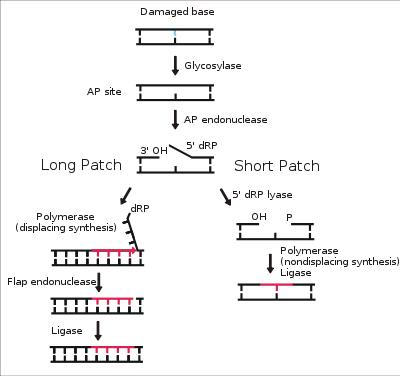
DNA ligase I functions to ligate single stranded DNA breaks in the final step of the base excision repair (BER) pathway.[14] The nitrogenous bases of DNA are commonly damaged by environmental hazards such as reactive oxygen species, toxins, and ionizing radiation. BER is the major repair pathway responsible for excising and replacing damaged bases. Ligase I is involved in the LP-BER pathway, whereas ligase III is involved in the major SN-BER pathway(2).[15] LP-BER proceeds in 4 catalytic steps. First, a DNA glycosylase cleaves the N-glycosidic bond, releasing the damaged base and creating an AP site– a site that lacks a purine or pyrimidine base. In the next step, an AP endonuclease creates a nick at the 5' end of the AP site, generating a hanging deoxyribose phosphate (dRP) residue in place of the AP site. DNA polymerase then synthesizes several new bases in the 5' to 3' direction, generating a hanging stretch of DNA with the dRP at its 5' end. It is at this step that SN-BER and LP-BER diverge in mechanism – in SNBER, only a single nucleotide is added and DNA Polymerase acts as a lyase to excise the AP site. In LP-BER, several bases are synthesized, generating a hanging flap of DNA, which is cleaved by a flap endonuclease. This leaves behind a nicked DNA strand that is sensed and ligated by DNA ligase.[14][15][16] The action of ligase I is stimulated by other LP-BER enzymes, particularly AP-endonuclease and DNA polymerase.[16]
Clinical significance
Mutations in LIG1 that lead to DNA ligase I deficiency result in immunodeficiency and increased sensitivity to DNA-damaging agents.[10]
There is only one confirmed case of a patient exhibiting ligase I deficiency, which resulted from an inherited mutant allele. The symptoms of this deficiency manifested as stunted growth and development and an immunodeficiency. A mouse model was made based on cell lines derived from the patient, confirming that the mutant ligase confers replication errors leading to genomic instability. Notably the mutant mice also showed increases in tumorigenesis.[8]
Ligase I has also been found to be upregulated in proliferating tumor cells, as opposed to benign tumor cell lines and normal human cells. Furthermore, it has been shown that inhibiting ligase I expression in these cells can have a cytotoxic effect, suggesting that ligase I inhibitors may be viable chemotherapeutic agents.[17]
Deficiencies in aprataxin, a phosphodiesterase responsible for reconditioning the DNA (after DNA ligase I aborts the adenylylated DNA intermediate), has been linked to neurodegeneration. This suggests that DNA is incapable of reentering the repair pathway without additional back-up machinery to correct for ligase errors.[13]
With the structure of DNA being well known and many of the components necessary for its manipulation, repair, and usage becoming identified and characterized, researchers are beginning to look into the development of nanoscopic machinery that would be incorporated into a living organism that would possess the ability to treat diseases, fight cancer, and release medications based on a biological stimulus provided by the organism to the nanosocpic machinery. DNA ligase would most likely have to be incorporated into such a machine.[18]
References
- GRCh38: Ensembl release 89: ENSG00000105486 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000056394 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Kresge N, Simoni RD, Hill RL (January 2007). "Insights into DNA Joining: I. Robert Lehman's work on DNA Ligase". Journal of Biological Chemistry. 282 (2): e1.
- Ferrari G, Rossi R, Arosio D, Vindigni A, Biamonti G, Montecucco A (September 2003). "Cell cycle-dependent phosphorylation of human DNA ligase I at the cyclin-dependent kinase sites". J. Biol. Chem. 278 (39): 37761–7. doi:10.1074/jbc.M304462200. PMID 12851383.
- Rossi R, Villa A, Negri C, Scovassi I, Ciarrocchi G, Biamonti G, Montecucco A (October 1999). "The replication factory targeting sequence/PCNA-binding site is required in G(1) to control the phosphorylation status of DNA ligase I". EMBO J. 18 (20): 5745–54. doi:10.1093/emboj/18.20.5745. PMC 1171641. PMID 10523317.
- Ellenberger T, Tomkinson AE (2008). "Eukaryotic DNA ligases: structural and functional insights". Annu. Rev. Biochem. 77: 313–38. doi:10.1146/annurev.biochem.77.061306.123941. PMC 2933818. PMID 18518823.
- Prigent C, Lasko DD, Kodama K, Woodgett JR, Lindahl T (August 1992). "Activation of mammalian DNA ligase I through phosphorylation by casein kinase II". EMBO J. 11 (8): 2925–33. doi:10.1002/j.1460-2075.1992.tb05362.x. PMC 556774. PMID 1639065.
- "Entrez Gene: LIG1 ligase I, DNA, ATP-dependent".
- Sriskanda V, Shuman S (January 1998). "Chlorella virus DNA ligase: nick recognition and mutational analysis". Nucleic Acids Res. 26 (2): 525–31. doi:10.1093/nar/26.2.525. PMC 147278. PMID 9421510.
- Taylor MR, Conrad JA, Wahl D, O'Brien PJ (July 2011). "Kinetic mechanism of human DNA ligase I reveals magnesium-dependent changes in the rate-limiting step that compromise ligation efficiency". J. Biol. Chem. 286 (26): 23054–62. doi:10.1074/jbc.M111.248831. PMC 3123073. PMID 21561855.
- Rass U, Ahel I, West SC (March 2007). "Actions of aprataxin in multiple DNA repair pathways". J. Biol. Chem. 282 (13): 9469–74. doi:10.1074/jbc.M611489200. PMID 17276982.
- Sattler U, Frit P, Salles B, Calsou P (April 2003). "Long-patch DNA repair synthesis during base excision repair in mammalian cells". EMBO Rep. 4 (4): 363–7. doi:10.1038/sj.embor.embor796. PMC 1319152. PMID 12671676.
- Hegde ML, Hazra TK, Mitra S (January 2008). "Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells". Cell Res. 18 (1): 27–47. doi:10.1038/cr.2008.8. PMC 2692221. PMID 18166975.
- Balakrishnan L, Brandt PD, Lindsey-Boltz LA, Sancar A, Bambara RA (May 2009). "Long patch base excision repair proceeds via coordinated stimulation of the multienzyme DNA repair complex". J. Biol. Chem. 284 (22): 15158–72. doi:10.1074/jbc.M109.000505. PMC 2685697. PMID 19329425.
- Sun D, Urrabaz R, Nguyen M, Marty J, Stringer S, Cruz E, Medina-Gundrum L, Weitman S (December 2001). "Elevated expression of DNA ligase I in human cancers". Clin. Cancer Res. 7 (12): 4143–8. PMID 11751514.
- Macdonald, Joanne. "Smart DNA: Programming the Molecule of Life for Work and Play [Preview]". scientificamerican. Retrieved 2013-02-22.
Further reading
- Leonhardt H, Cardoso MC (1996). "Targeting and association of proteins with functional domains in the nucleus: the insoluble solution". Int. Rev. Cytol. International Review of Cytology. 162B: 303–35. doi:10.1016/S0074-7696(08)62620-0. ISBN 9780123645661. PMID 8557490.
- Tomkinson AE, Mackey ZB (1998). "Structure and function of mammalian DNA ligases". Mutat. Res. 407 (1): 1–9. doi:10.1016/s0921-8777(97)00050-5. PMID 9539976.
- Perrigot M, Pierrot-Deseilligny E, Bussel B, Held JP (1976). "[Paralysis following Dimer X radiculography]". La Nouvelle Presse Médicale. 5 (17): 1120–2. PMID 934827.
- Webster AD, Barnes DE, Arlett CF, et al. (1992). "Growth retardation and immunodeficiency in a patient with mutations in the DNA ligase I gene". Lancet. 339 (8808): 1508–9. doi:10.1016/0140-6736(92)91266-B. PMID 1351188.
- Barnes DE, Tomkinson AE, Lehmann AR, et al. (1992). "Mutations in the DNA ligase I gene of an individual with immunodeficiencies and cellular hypersensitivity to DNA-damaging agents". Cell. 69 (3): 495–503. doi:10.1016/0092-8674(92)90450-Q. PMID 1581963.
- Barnes DE, Kodama K, Tynan K, et al. (1992). "Assignment of the gene encoding DNA ligase I to human chromosome 19q13.2-13.3". Genomics. 12 (1): 164–6. doi:10.1016/0888-7543(92)90422-O. PMID 1733856.
- Petrini JH, Huwiler KG, Weaver DT (1991). "A wild-type DNA ligase I gene is expressed in Bloom's syndrome cells". Proc. Natl. Acad. Sci. U.S.A. 88 (17): 7615–9. doi:10.1073/pnas.88.17.7615. PMC 52352. PMID 1881902.
- Lasko DD, Tomkinson AE, Lindahl T (1990). "Mammalian DNA ligases. Biosynthesis and intracellular localization of DNA ligase I.". J. Biol. Chem. 265 (21): 12618–22. PMID 2197279.
- Barnes DE, Johnston LH, Kodama K, et al. (1990). "Human DNA ligase I cDNA: cloning and functional expression in Saccharomyces cerevisiae". Proc. Natl. Acad. Sci. U.S.A. 87 (17): 6679–83. doi:10.1073/pnas.87.17.6679. PMC 54600. PMID 2204063.
- Montecucco A, Savini E, Weighardt F, et al. (1996). "The N-terminal domain of human DNA ligase I contains the nuclear localization signal and directs the enzyme to sites of DNA replication". EMBO J. 14 (21): 5379–86. doi:10.1002/j.1460-2075.1995.tb00222.x. PMC 394647. PMID 7489727.
- Maruyama K, Sugano S (1994). "Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides". Gene. 138 (1–2): 171–4. doi:10.1016/0378-1119(94)90802-8. PMID 8125298.
- Trask B, Fertitta A, Christensen M, et al. (1993). "Fluorescence in situ hybridization mapping of human chromosome 19: cytogenetic band location of 540 cosmids and 70 genes or DNA markers". Genomics. 15 (1): 133–45. doi:10.1006/geno.1993.1021. PMID 8432525.
- Petrini JH, Walsh ME, DiMare C, et al. (1996). "Isolation and characterization of the human MRE11 homologue". Genomics. 29 (1): 80–6. doi:10.1006/geno.1995.1217. PMID 8530104.
- Bentley D, Selfridge J, Millar JK, et al. (1996). "DNA ligase I is required for fetal liver erythropoiesis but is not essential for mammalian cell viability". Nat. Genet. 13 (4): 489–91. doi:10.1038/ng0896-489. PMID 8696349.
- Suzuki Y, Yoshitomo-Nakagawa K, Maruyama K, et al. (1997). "Construction and characterization of a full length-enriched and a 5'-end-enriched cDNA library". Gene. 200 (1–2): 149–56. doi:10.1016/S0378-1119(97)00411-3. PMID 9373149.
- Rossi R, Villa A, Negri C, et al. (1999). "The replication factory targeting sequence/PCNA-binding site is required in G(1) to control the phosphorylation status of DNA ligase I." EMBO J. 18 (20): 5745–54. doi:10.1093/emboj/18.20.5745. PMC 1171641. PMID 10523317.
- Matsumoto Y, Kim K, Hurwitz J, et al. (1999). "Reconstitution of proliferating cell nuclear antigen-dependent repair of apurinic/apyrimidinic sites with purified human proteins". J. Biol. Chem. 274 (47): 33703–8. doi:10.1074/jbc.274.47.33703. PMID 10559261.
- Vispé S, Satoh MS (2000). "DNA repair patch-mediated double strand DNA break formation in human cells". J. Biol. Chem. 275 (35): 27386–92. doi:10.1074/jbc.M003126200. PMID 10827190.
External links
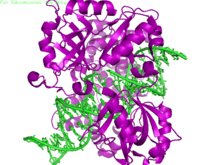
- Overview of all the structural information available in the PDB for UniProt: P18858 (DNA ligase 1) at the PDBe-KB.
PDB gallery | |
|---|---|
|