Juliá–Colonna epoxidation
The Juliá–Colonna epoxidation is an asymmetric poly-leucine catalyzed nucleophilic epoxidation of electron deficient olefins in a triphasic system. The reaction was reported by Sebastian Juliá at the Chemical Institute of Sarriá in 1980,[1] with further elaboration by both Juliá and Stefano Colonna (Istituto di Chimica Industriale dell'Università, Milan, Italy).[2]
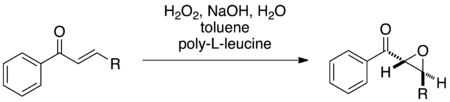 The Juliá–Colonna Epoxidation of a chalcone proceeds with poly-L-leucine and hydrogen peroxide in generic triphasic conditions. Image adapted from Juliá et al.[2]
The Juliá–Colonna Epoxidation of a chalcone proceeds with poly-L-leucine and hydrogen peroxide in generic triphasic conditions. Image adapted from Juliá et al.[2]
In the original triphasic protocol, the chalcone substrate is soluble in the organic phase, generally toluene or carbon tetrachloride. The alkaline hydrogen peroxide oxidant is soluble primarily in the aqueous phase, and the reaction occurs at the insoluble polymer layer at the interface of the two phases. Alternative biphasic and monophasic protocols have been developed with increased substrate accessibility and reaction rate.[3][4]
The efficient enantioselective catalytic epoxidation under mild conditions is of great synthetic utility. Not only are epoxides effective synthons for a range of transformations, they have a significant presence in natural products structures. Furthermore, the reaction has been effectively scaled up to industrially useful levels, with work conducted notably by Bayer and Evonik. Finally, the enzyme-like activity of the poly-amino acid segments is suggestive of a role of the reaction in the prebiotic origin of life.[5][6]
Reaction mechanism
The Juliá–Colonna epoxidation is an asymmetric nucleophilic epoxidation of electron-deficient olefins such as α,β-unsaturated ketones. The general mechanism shown in Figure 2 applies to all nucleophilic epoxidations but is controlled in this reaction by the poly-leucine catalyst.
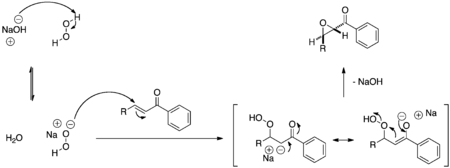
The hydroperoxide anion and chalcone assemble in a complex with the poly-leucine catalyst before reacting to form a peroxide enolate intermediate. The intermediate subsequently closes, as controlled by the catalyst structure, to form the epoxide product stereoselectively.
Ternary complex formation
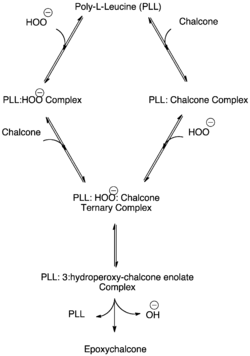
The poly-leucine strands demonstrate enzyme-like kinetics with a first-order dependence on and eventual saturation with both the hydroperoxide anion (KM= 30 mM) and the olefin substrate (KM=110 mM.) Kinetic study suggests that the reaction proceeds by random steady-state formation of a ternary (polyleucine+hydroperoxide anion+olefin) complex. Both substrates must bind prior to reaction, and while either may bind first, initial hydroperoxide binding is kinetically preferred. The rapid equilibrium enabling complex formation is followed by the rate-limiting formation of the peroxide enolate (Figure 3).[5][8]
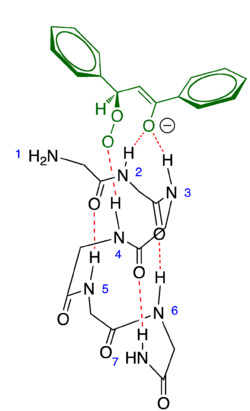
Mechanistic origin of stereoselectivity
All of the reactants associate with the polyleucine catalyst prior to reaction to form the hydroperoxide enolate intermediate. The catalyst orients the reactants and, even more significantly, the peroxide enolate intermediate by a series of hydrogen bonding interactions with the four N-terminal amino groups in the poly-leucine α-helix. While other models have been proposed,[9] computations by Kelly et al. have suggested that the NH-2, NH-3, and NH-4 form an isosceles triangle available for hydrogen bonding as an intermediate-stabilizing oxyanion hole. While olefin binding to either the endo or exo face of the helix is sterically allowed, only endo binding orients the NH-4 group to bind with the hydroperoxide moiety allowing for hydroxide displacement in the final reaction step (Figure 4).[7]
Catalyst
Poly-amino acid selection
Enantioselectivity is maximized by poly-amino acid sequences containing the greatest α-helical content; these include poly-leucine and poly-alanine.[1] Both poly-L- and poly-D-amino acids are available and cause the opposite stereoinduction.[10]
Catalyst generation
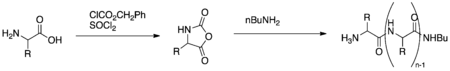
The original poly-leucine catalysts were formed by reacting leucine-N-carboxyanhydrides with an initiator such as an amine, an alcohol or water (Figure 5).[2] In triphasic systems, the polymer catalyst must be soaked in the organic solvent and peroxide solution to generate a gel prior to reaction.[11] – Especially in biphasic systems, reaction time may be reduced and enantioselectivity increased by activating the catalyst with NaOH prior to reaction. Furthermore, in biphasic systems the polymer may be immobilized on polystyrene, polyethylene glycol (PEG), or silica gel and formed into a paste.[4]
Catalyst secondary structure
The active component of the catalyst assumes an α-helical structure where the four to five N-terminal residues are actively involved in catalysis. While active catalysts have been generated from scalemic leucine, consistent enantiomeric content must be maintained through the N-terminal region to give appropriate handedness to the structure.[10] While the greatest enantioselectivity was originally observed when n=30 residues,[2] a 10-mer Leucine polypeptide is of sufficient length to provide significant enantioselectivity[10] Following improvement of the original procedure, greater enantioselectivity has been observed for lower molecular weight polymers, presumably due to the greater number of N-termini available per mass used.[4]
Scope
The Juliá–Colonna epoxidation of electron-deficient olefins was originally demonstrated with chalcones, but it was soon extended to other systems with electron withdrawing moieties such as α,β-unsaturated ketones, esters, and amides.[1][2] The reaction has also demonstrated efficiency with sulfone substrates, and the scope of the reaction is being expanded with further methdological investigation.[12]
Several classes of substrates, however, are not suitable for the Juliá–Colonna Epoxidation. These include:[10]
- compounds sensitive to hydroxide.
- compounds with acidic protons on the α or α’ positions.
- electron rich olefins.
The nucleophilic epoxidation is naturally complementary in scope to electrophilic epoxidations such as the Sharpless epoxidation and Jacobsen epoxidation.
Stereoselectivity
Catalyst structure
The stereoinduction of the Juliá–Colonna epoxidation is dependent on the α-helical secondary structure of the poly-leucine catalyst. While the consistent stereochemistry of the N-terminal amino acids is necessary for this induction, even a 10-mer leucine polypeptide is of sufficient length to provide significant enantioselectivity.[10]
Chiral amplification by scalemic catalysts
This dependence solely on the N-terminal region of the helix is most pronounced in enantioselective stereoinduction by scalemic catalysts. Even a 40% enantiomeric excess of L vs. D-leucine in catalyst formation can yield the same enantiomeric enriched epoxide as the enantiopure catalyst. The relationship between catalyst and product enantiopurity can be closely approximated with a Bernoullian statistical model: een=(Ln-Dn)/(Ln+Dn) where L and D are the proportions of L- and D-leucine used to generate the catalytic polymers and n is the length of the catalytic component.[5][6]
Chiral amino acids, including leucine, have been generated in electrical discharge experiments designed to mimic the prebiotic conditions on Earth, and they have been found in scalemic mixtures in meteorites. It has been suggested that poly-amino acid fragments analogous to the Juliá–Colonna catalyst may have been initiated by imidazole or cyanide derivatives, and the resulting fragments may have played a catalytic role in the origin of enantiomeric enrichment ubiquitous in life today.[5]
Variations
Silica-grafted catalysts
Silica-grafted polyleucine has been shown to effectively catalyze epoxidation of α,β-unsaturated aromatic ketones. The silica graft allows for the catalyst to be easily recovered with only mild loss of activity and is particularly useful for scale-up reactions.[13]
Biphasic (non-aqueous) reaction conditions
For the alternative biphasic protocol, the olefin substrate is dissolved in tetrahydrofuran (THF) along with the urea hydrogen peroxide (UHP) oxidant and a tertiary amine base such as 8-diazabicyclo[5.4.0]undec-7-ene (DBU.) The immobilized polymer catalyst forms a paste which serves as the reaction site. The two phase reaction conditions extended the range of enones to which the reaction could be applied.[3]
Monophasic reaction conditions with PEG-immobilized polyleucine
A soluble initiator O,O′-bis(2-aminoethyl)polyethylene glycol (diaminoPEG) for poly-leucine assembly was utilized to generate a THF-soluble triblock polymer. Utilization of this catalyst in homogeneous reaction conditions enabled marked extension of the methodology to α,β-unsaturated ketones, dienes, and bis-dienes.[4]
Phase transfer co-catalysis
Addition of tetrabutylammonium bromide as a phase transfer catalyst dramatically increases the rate of reaction. The co-catalyst is presumed to increase the concentration of the peroxide oxidant in the organic phase enabling more efficient access to the reactive ternary complex.[14] These conditions were developed for application to two phase systems but also function for three phase systems and have been utilized up to the 100g scale[5][12]
Scale-up
Immobilized catalysts have been used in membrane reactors and are being investigated for application to continuous flow fixed bed reactors.[11]
Applications to synthesis
Total synthesis of Diltiazem
Adger et al. utilized the biphasic Juliá–Colonna epoxidation with immobilized poly-L-leucine (I-PLL) and urea hydrogen peroxide (UHP), and 8-diazabicyclo[5.4.0]undec-7-ene (DBU) as the key step in the efficient synthesis of Diltiazem (Figure 6.) Diltiazem is a commercially available pharmaceutical which acts as a calcium channel blocker.[11]
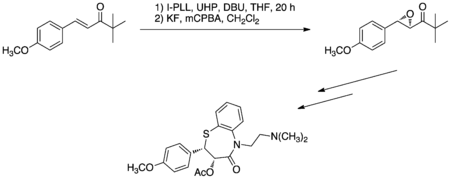
Total synthesis of (+)-clausenamide
Cappi et al. utilized the Juliá–Colonna epoxidation with PEG-immobilized poly-L-leucine (PEG-PLL) and DABCO hydrogen peroxide (DABCO-H2O2) or urea hydrogen peroxide (UHP) in a miniature fixed-bed continuous flow reactor system (Figure 7.) This protocol was exploited to synthesize (+)-clausenamide as a proof of concept in the development of the novel reaction protocol; (+)-clausenamide exhibits anti-amnesiac and hepatoprotective activity.[15]
-Clausenamide7.png)
Total synthesis of (+)-goniotriol 7, (+)-goniofufurone 8, (+)-8-acetylgoniotriol 9 and gonio-pypyrone
Chen et al. utilized the biphasic Juliá–Colonna Epoxidation protocol with urea hydrogen peroxide (UHP), poly-L-leucine (PLL), and 8-diazabicyclo[5.4.0]undec-7-ene (DBU) as a key step in the synthesis of a family of styryl lactones isolated from Goniothalamus giganteus (Figure 8.) These compounds, including (+)-goniotriol 7, (+)-goniofufurone 8, (+)-8-acetylgoniotriol 9 and gonio-pypyrone, have demonstrated cytotoxic activity against human tumor cells.[16]
-goniotriol.png)
See also
- Prilezhaev reaction
- Jørgensen epoxidation
- Asymmetric nucleophilic epoxidation
References
- Juliá, S. N.; Masana, J.; Vega, J. C. (1980). ""Synthetic Enzymes". Highly Stereoselective Epoxidation of Chalcone in a Triphasic Toluene-Water-Poly\(S)-alanine] System". Angewandte Chemie International Edition in English. 19 (11): 929. doi:10.1002/anie.198009291.
- Juliá, Sebastián; Guixer, Joan; Masana, Jaume; Rocas, José; Colonna, Stefano; Annuziata, Rita; Molinari, Henriette (1982). "Synthetic enzymes. Part 2. Catalytic asymmetric epoxidation by means of polyamino-acids in a triphase system". J. Chem. Soc., Perkin Trans. 1: 1317. doi:10.1039/P19820001317.
- Allen, Joanne V.; Bergeron, Sophie; Griffiths, Matthew J.; Mukherjee, Shubhasish; Roberts, Stanley M.; Williamson, Natalie M.; Wu, L. Eduardo (1998). "Juliá–Colonna asymmetric epoxidation reactions under non-aqueous conditions: rapid, highly regio- and stereo-selective transformations using a cheap, recyclable catalyst". J. Chem. Soc., Perkin Trans. 1 (19): 3171. doi:10.1039/A805407J.
- Flood, Robert W.; Geller, Thomas P.; Petty, Sarah A.; Roberts, Stanley M.; Skidmore, John; Volk, Martin (2001). "Efficient Asymmetric Epoxidation of α,β-Unsaturated Ketones Using a Soluble Triblock Polyethylene Glycol−Polyamino Acid Catalyst". Org. Lett. 3 (5): 683. doi:10.1021/ol007005l.
- Carrea, G; Colonna, S; Kelly, D; Lazcano, A; Ottolina, G; Roberts, S (2005). "Polyamino acids as synthetic enzymes: mechanism, applications and relevance to prebiotic catalysis". Trends in Biotechnology. 23 (10): 507. doi:10.1016/j.tibtech.2005.07.010.
- Kelly, David R.; Meek, Alastair; Roberts, Stanley M. (2004). "Chiral amplification by polypeptides and its relevance to prebiotic catalysis". Chem. Comm. (18): 2021. doi:10.1039/B404379K.
- Kelly, D. R.; Roberts, S. M., The mechanism of polyleucine catalysed asymmetric epoxidation". Chem. Comm. 2004, (18), 2018-2020. doi:10.1039/B404390C
- Carrea, G.; Colonna, S.; Meek, A. D.; Ottolina, G.; Roberts, S. M., "Kinetics of chalcone oxidation by peroxide anion catalysed by poly-L-leucine". Chem. Comm. 2004, (12), 1412-1413. doi:10.1039/B401497A
- Berkessel, A.; Gasch, N.; Glaubitz, K.; Koch, C., "Highly enantioselective enone epoxidation catalyzed by short solid phase-bound peptides: Dominant role of peptide helicity". Org. Lett. 2001, 3 (24), 3839–3842. doi:10.1021/ol0166451
- Bentley, P. A.; Cappi, M. W.; Flood, R. W.; Roberts, S. M.; Smith, J. A., Towards a mechanistic insight into the Julia-Colonna asymmetric epoxidation of α,β-unsaturated ketones using discrete lengths of poly-leucine. Tetrahedron Lett. 1998, 39 (50), 9297–9300. doi:10.1016/S0040-4039(98)02090-5
- Adger, B. M.; Barkley, J. V.; Bergeron, S.; Cappi, M. W.; Flowerdew, B. E.; Jackson, M. P.; McCague, R.; Nugent, T. C.; Roberts, S. M., "Improved procedure for Julia–Colonna asymmetric epoxidation of α,β-unsaturated ketones: total synthesis of diltiazem and Taxol (TM) side-chain". J. Chem. Soc.-Perkin Trans. 1 1997, (23), 3501–3507. doi:10.1039/A704413E
- Lopez-Pedrosa, J. M.; Pitts, M. R.; Roberts, S. M.; Saminathan, S.; Whittall, J., "Asymmetric epoxidation of some arylalkenyl sulfones using a modified Julia–Colonna procedure". Tetrahedron Lett. 2004, 45 (26), 5073–5075. doi:10.1016/j.tetlet.2004.04.190
- Yi, H.; Zou, G.; Li, Q.; Chen, Q.; Tang, J.; He, M. Y., "Asymmetric epoxidation of alpha,beta-unsaturated ketones catalyzed by silica-grafted poly-(L)-leucine catalysts". Tetrahedron Lett. 2005, 46 (34), 5665–5668. doi:10.1016/j.tetlet.2005.06.096
- Geller, T.; Gerlach, A.; Kruger, C. M.; Militzer, H. C., "Novel conditions for the Julia–Colonna epoxidation reaction providing efficient access to chiral, nonracemic epoxides". Tetrahedron Lett. 2004, 45 (26), 5065–5067. doi:10.1016/j.tetlet.2004.04.188
- Cappi, M. W.; Chen, W. P.; Flood, R. W.; Liao, Y. W.; Roberts, S. M.; Skidmore, J.; Smith, J. A.; Williamson, N. M., "New procedures for the –Colonna asymmetric epoxidation: synthesis of (+)-clausenamide". Chem. Comm. 1998, (10), 1159-1160. doi:10.1039/A801450G
- Chen, W. P.; Roberts, S. M., "Julia–Colonna asymmetric epoxidation of furyl styryl ketone as a route to intermediates to naturally-occurring styryl lactones". J. Chem. Soc.-Perkin Trans. 1 1999, (2), 103–105. doi:10.1039/A808436J