Gaucher's disease
Gaucher's disease or Gaucher disease (/ɡoʊˈʃeɪ/) (GD) is a genetic disorder in which glucocerebroside (a sphingolipid, also known as glucosylceramide) accumulates in cells and certain organs. The disorder is characterized by bruising, fatigue, anemia, low blood platelet count and enlargement of the liver and spleen, and is caused by a hereditary deficiency of the enzyme glucocerebrosidase (also known as glucosylceramidase), which acts on glucocerebroside. When the enzyme is defective, glucocerebroside accumulates, particularly in white blood cells and especially in macrophages (mononuclear leukocytes). Glucocerebroside can collect in the spleen, liver, kidneys, lungs, brain, and bone marrow.
| Gaucher's disease | |
|---|---|
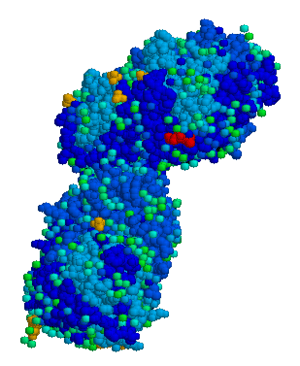 | |
| Acid beta-glucosidase | |
| Specialty | Endocrinology, neurology |
Manifestations may include enlarged spleen and liver, liver malfunction, skeletal disorders or bone lesions that may be painful, severe neurological complications, swelling of lymph nodes and (occasionally) adjacent joints, distended abdomen, a brownish tint to the skin, anemia, low blood platelet count, and yellow fatty deposits on the white of the eye (sclera). Persons seriously affected may also be more susceptible to infection. Some forms of Gaucher's disease may be treated with enzyme replacement therapy.
The disease is caused by a recessive mutation in the GBA gene located on chromosome 1 and affects both males and females. About one in 100 people in the United States are carriers of the most common type of Gaucher disease. The carrier rate among Ashkenazi Jews is 8.9% while the birth incidence is one in 450.[1]
Gaucher's disease is the most common of the lysosomal storage diseases.[2] It is a form of sphingolipidosis (a subgroup of lysosomal storage diseases), as it involves dysfunctional metabolism of sphingolipids.[3]
The disease is named after the French physician Philippe Gaucher, who originally described it in 1882.[4]
Signs and symptoms
- Painless hepatomegaly and splenomegaly: the size of the spleen can be 1500–3000 mg, as opposed to the normal size of 50–200 mg. Splenomegaly may decrease the affected individual's capacity for eating by exerting pressure on the stomach. While painless, enlargement of spleen increases the risk of splenic rupture.
- Hypersplenism and pancytopenia, the rapid and premature destruction of blood cells, leads to anemia, neutropenia, leukopenia, and thrombocytopenia (with an increased risk of infection and bleeding).
- Cirrhosis of the liver is rare.
- Severe pain associated with joints and bones occurs, frequently presenting in hips and knees.
- Neurological symptoms occur only in some types of Gaucher's (see below):
- Type I: impaired olfaction and cognition
- Type II: serious convulsions, hypertonia, intellectual disability, and apnea
- Type III: muscle twitches known as myoclonus, convulsions, dementia, and ocular muscle apraxia
- Parkinson's disease is recognized as being more common in Gaucher's disease patients and their heterozygous carrier relatives.[5]
- Osteoporosis: 75% of patients develop visible bony abnormalities due to the accumulated glucosylceramide. A deformity of the distal femur in the shape of an Erlenmeyer flask is commonly described.
- Yellowish-brown skin pigmentation
Genetics
The three types of Gaucher's disease are autosomal recessive. Both parents must be carriers for a child to be affected. If both parents are carriers, the chance of the disease is one in four, or 25%, with each pregnancy for an affected child. Genetic counseling and genetic testing are recommended for families who may be carriers of mutations.
Each type has been linked to particular mutations. In all, about 80 known GBA gene mutations are grouped into three main types:[6]
- Type I (N370S homozygote), the most common, also called the "non-neuropathic" type occurs mainly in Ashkenazi Jews, at 100 times the occurrence in the general populace. The median age at diagnosis is 28 years of age,[7] and life expectancy is mildly decreased.[8]
- Type II (one or two alleles L444P) is characterized by neurological problems in small children. The enzyme is hardly released into the lysosomes. Prognosis is poor: most die before the age of three.
- Type III (also one or two copies of L444P, possibly delayed by protective polymorphisms) occurs in Swedish patients from the Norrbotten region.[9] This group develops the disease somewhat later, but most die before their 30th birthday.
The Gaucher-causing mutations may have entered the Ashkenazi Jewish gene pool in the early Middle Ages (48–55 generations ago).[10]
Pathophysiology
The disease is caused by a defect in housekeeping gene for lysosomal glucocerebrosidase (also known as beta-glucosidase, EC 3.2.1.45, PDB: 1OGS) on the first chromosome (1q22). The enzyme is a 55.6-kilodalton, 497-amino acid-long protein that catalyses the breakdown of glucocerebroside, a cell membrane constituent of red and white blood cells. In Gaucher disease, the enzyme is unable to function correctly and glucocerebroside accumulates. The macrophages that clear these cells are unable to eliminate the waste product, which accumulates in fibrils, and turn into 'Gaucher cells', which appear on light microscopy to resemble crumpled-up paper.[3]
The exact mechanism of neurotoxicity is not understood, but it is thought to involve a reaction to glucosylsphingosine.[3]
Different mutations in the GBA (beta-glucosidase) gene determine the remaining activity of the enzyme. In type I, there is some residual activity of the enzyme, accounting for the lack of neuropathology in this type.[3] Although there is some correlation between genotype and phenotype, neither the amount of stored lipids, nor the residual enzyme activity correlates well with disease symptoms.[11] This circumstance has called for alternative explanations accounting for disease symptoms including
- jamming of the endo/lysosomal system[12]
- ER stress[13]
- altered lipid composition of membranes throughout the cell, including the plasma membrane,[14] and consequent changes in the dynamic and signaling properties of the cell membrane[15]
- inflammation caused by cytokine secretion as a result of sphingolipid accumulation, and neurodegeneration caused by the accumulation of glucosylsphingosine, a neurotoxin[16]
Heterozygotes for particular acid beta-glucosidase mutations carry about a five-fold risk of developing Parkinson's disease, making this the most common known genetic risk factor for Parkinson's.[17][18]
Cancer risk may be increased, particularly myeloma.[19][20][21] This is thought to be due to accumulation of glucosylceramide and complex glycosphingolipids.[22]
The role of inflammatory processes in Gaucher disease is poorly elucidated. However, sphingolipids are known to participate in inflammation and apoptosis, and markers of macrophage activation are elevated in people with Gaucher disease. These markers include angiotensin-converting enzyme, cathepsin S, chitotriosidase, and CCL18 in the blood plasma; and tumor necrosis factor alpha in splenic Gaucher cells (engorged macrophages).[3]
Diagnosis
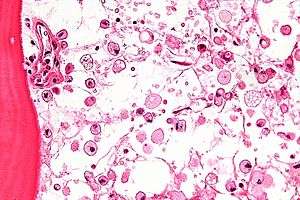
Gaucher disease is suggested based on the overall clinical picture. Initial laboratory testing may include enzyme testing. As a result, lower than 15% of mean normal activity is considered to be diagnostic.[23] Decreased enzyme levels will often be confirmed by genetic testing. Numerous different mutations occur; sequencing of the beta-glucosidase gene is sometimes necessary to confirm the diagnosis. Prenatal diagnosis is available and is useful when a known genetic risk factor is present.
A diagnosis can also be implied by biochemical abnormalities such as high alkaline phosphatase, angiotensin-converting enzyme, and immunoglobulin levels, or by cell analysis showing "crinkled paper" cytoplasm and glycolipid-laden macrophages.
Some lysosomal enzymes are elevated, including tartrate-resistant acid phosphatase, hexosaminidase, and a human chitinase, chitotriosidase. This latter enzyme has proved to be very useful for monitoring Gaucher's disease activity in response to treatment, and may reflect the severity of the disease
Classification
Gaucher's disease (GD) has four common clinical subtypes.[24][25] These subtypes have come under some criticism for not taking account of the full spectrum of observable symptoms (the phenotypes[26]). Also, compound heterozygous variations occur which considerably increase the complexity of predicting disease course.
GD type I (non-neuropathic) is the most common and least severe form of the disease. Symptoms may begin early in life or in adulthood and mainly affect the liver, spleen, and bone. Enlarged liver and grossly enlarged spleen (together hepatosplenomegaly) are common;[3] the spleen can rupture and cause additional complications. Skeletal weakness and bone disease may be extensive.[3] Spleen enlargement and bone marrow replacement cause anemia, thrombocytopenia, and leukopenia. The brain and nervous system are not affected pathologically,[3] but lung and, rarely, kidney impairment may occur. Patients in this group usually bruise easily (due to low levels of platelets) and experience fatigue due to low numbers of red blood cells. Depending on disease onset and severity, type I patients may live well into adulthood. The range and severity of symptoms can vary dramatically between patients.
GD type II (acute infantile neuropathic) typically begins within 6 months of birth and has an incidence rate around one 1 in 100,000 live births. Symptoms include an enlarged liver and spleen, extensive and progressive brain damage, eye movement disorders, spasticity, seizures, limb rigidity, and a poor ability to suck and swallow. Affected children usually die by age two.
GD type III (chronic neuropathic) can begin at any time in childhood or even in adulthood, and occurs in about one in 100,000 live births. It is characterized by slowly progressive, but milder neurologic symptoms compared to the acute or type II version. Major symptoms include an enlarged spleen and/or liver, seizures, poor coordination, skeletal irregularities, eye movement disorders, blood disorders including anemia, and respiratory problems. Patients often live into their early teen years and adulthood.[27]
Treatment
For those with type-I and most type-III, enzyme replacement treatment with intravenous recombinant glucocerebrosidase can decrease liver and spleen size, reduce skeletal abnormalities, and reverse other manifestations.[16][28] This treatment costs about US$200,000 annually for a single person and should be continued for life. The rarity of the disease means dose-finding studies have been difficult to conduct, so controversy remains over the optimal dose and dosing frequency.[7] Due to the low incidence, this has become an orphan drug in many countries, meaning a government recognizes and accommodates the financial constraints that limit research into drugs that address a small population.
The first drug for Gaucher's was alglucerase (Ceredase), which was a version of glucocerebrosidase that was harvested from human placental tissue and then modified with enzymes.[29] It was approved by the FDA in 1991[30] and has been withdrawn from the market[31][32] due to the approval of similar drugs made with recombinant DNA technology instead of being harvested from tissue; drugs made recombinantly are preferable, since there is no concern about diseases being transmitted from the tissue used in harvesting, there are fewer risks of variations in enzyme structure from batch to batch, and they are less expensive to manufacture.[29]
Available recombinant glucocerebrosidases are:[16]
- Imiglucerase (approved in 1995)[29]
- Velaglucerase (approved in 2010)[33]
- Taliglucerase alfa (Elelyso) (approved in 2012)[34]
Miglustat is a small molecule, orally available drug that was first approved for Gaucher's Disease in Europe in 2002.[35] It works by preventing the formation of glucocerebroside, the substance that builds up and causes harm in Gaucher's. This approach is called substrate reduction therapy.[36]
Eliglustat (Cerdelga) (approved in 2014) [37] is also a small molecule. The compound is believed to work by inhibition of glucosylceramide synthase.
Epidemiology
The National Gaucher Foundation (United States) states the incidence of Gaucher's disease is about one in 20,000 live births.[38] Around one in 100 people in the general US population is a carrier for type I Gaucher's disease, giving a prevalence of one in 40,000.[39] Among Ashkenazi Jews, the rate of carriers is considerably higher, at roughly one in 15.[39]
Type II Gaucher's disease shows no particular preference for any ethnic group.
Type III Gaucher's disease is especially common in the population of the northern Swedish region of Norrbotten, where the incidence of the disease is one in 50,000.[40]
History
The disease was first recognized by the French doctor Philippe Gaucher, who originally described it in 1882 and lent his name to the condition.[4] In 1902, its mode of inheritance was discovered by Nathan Brill.[3] The neuronal damage associated with the disease was discovered in the 1920s, and the biochemical basis for the disease was elucidated in the 1960s by Roscoe Brady.[3][41] The first effective treatment for the disease, the drug alglucerase (Ceredase), was approved by the FDA in April 1991. An improved drug, imiglucerase (Cerezyme), was approved by the FDA in May 1994 and has replaced the use of Ceredase.
October is National Gaucher's Disease Awareness Month in the United States.
Prominent people with disease
- Wallace Chapman; New Zealand radio and television personality[42]
Gallery
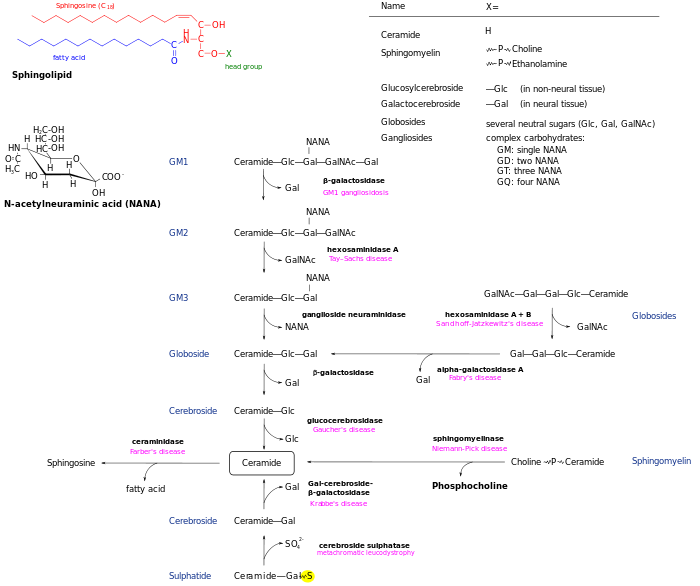 Sphingolipidoses
Sphingolipidoses
See also
- Niemann–Pick disease
- Fabry disease
- Tay–Sachs disease
- Krabbe disease
- Metachromatic leukodystrophy
- Medical genetics of Ashkenazi Jews
- List of radiographic findings associated with cutaneous conditions
References
- Zimran A, Gelbart T, Westwood B, Grabowski GA, Beutler E (October 1991). "High frequency of the Gaucher disease mutation at nucleotide 1226 among Ashkenazi Jews". American Journal of Human Genetics. 49 (4): 855–9. PMC 1683177. PMID 1897529.
- James WD, Elston DM, Berger TG, Andrews GC (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. p. 536. ISBN 978-0-7216-2921-6. OCLC 663444979.
- Dandana A, Ben Khelifa S, Chahed H, Miled A, Ferchichi S (2016). "Gaucher Disease: Clinical, Biological and Therapeutic Aspects". Pathobiology. 83 (1): 13–23. doi:10.1159/000440865. PMID 26588331.
- Gaucher PCE (1882). De l'epithelioma primitif de la rate, hypertrophie idiopathique de la rate sans leucemie [Primary epithelioma of the spleen, idiopathic hypertrophy of the spleen without leukemia] (academic thesis) (in French). Paris, France.
- McNeill A, Duran R, Hughes DA, Mehta A, Schapira AH (August 2012). "A clinical and family history study of Parkinson's disease in heterozygous glucocerebrosidase mutation carriers". Journal of Neurology, Neurosurgery, and Psychiatry. 83 (8): 853–4. doi:10.1136/jnnp-2012-302402. PMC 3927562. PMID 22577228.
- Online Mendelian Inheritance in Man (OMIM): Gluosidase, beta, acid; GBA - 606463
- Grabowski GA (October 2008). "Phenotype, diagnosis, and treatment of Gaucher's disease". Lancet. 372 (9645): 1263–71. doi:10.1016/S0140-6736(08)61522-6. PMID 19094956.
- Weinreb NJ, Deegan P, Kacena KA, Mistry P, Pastores GM, Velentgas P, vom Dahl S (December 2008). "Life expectancy in Gaucher disease type 1". American Journal of Hematology. 83 (12): 896–900. doi:10.1002/ajh.21305. PMC 3743399. PMID 18980271.
- Dahl N, Lagerström M, Erikson A, Pettersson U (August 1990). "Gaucher disease type III (Norrbottnian type) is caused by a single mutation in exon 10 of the glucocerebrosidase gene". American Journal of Human Genetics. 47 (2): 275–8. PMC 1683716. PMID 2378352.
- Diaz GA, Gelb BD, Risch N, Nygaard TG, Frisch A, Cohen IJ, Miranda CS, Amaral O, Maire I, Poenaru L, Caillaud C, Weizberg M, Mistry P, Desnick RJ (June 2000). "Gaucher disease: the origins of the Ashkenazi Jewish N370S and 84GG acid beta-glucosidase mutations". American Journal of Human Genetics. 66 (6): 1821–32. doi:10.1086/302946. PMC 1378046. PMID 10777718.
- Sidransky E (October 2012). "Gaucher disease: insights from a rare Mendelian disorder". Discovery Medicine. 14 (77): 273–81. PMC 4141347. PMID 23114583.
- Simons K, Gruenberg J (November 2000). "Jamming the endosomal system: lipid rafts and lysosomal storage diseases". Trends in Cell Biology. 10 (11): 459–62. doi:10.1016/S0962-8924(00)01847-X. PMID 11050411.
- Westbroek W, Gustafson AM, Sidransky E (September 2011). "Exploring the link between glucocerebrosidase mutations and parkinsonism". Trends in Molecular Medicine. 17 (9): 485–93. doi:10.1016/j.molmed.2011.05.003. PMC 3351003. PMID 21723784.
- Hein LK, Meikle PJ, Hopwood JJ, Fuller M (December 2007). "Secondary sphingolipid accumulation in a macrophage model of Gaucher disease". Molecular Genetics and Metabolism. 92 (4): 336–45. doi:10.1016/j.ymgme.2007.08.001. PMID 17881272.
- Batta G, Soltész L, Kovács T, Bozó T, Mészár Z, Kellermayer M, Szöllősi J, Nagy P (January 2018). "Alterations in the properties of the cell membrane due to glycosphingolipid accumulation in a model of Gaucher disease". Scientific Reports. 8 (1): 157. Bibcode:2018NatSR...8..157B. doi:10.1038/s41598-017-18405-8. PMC 5760709. PMID 29317695.
- Grabowski GA (2012). "Gaucher disease and other storage disorders". Hematology. American Society of Hematology. Education Program. 2012: 13–8. doi:10.1182/asheducation.v2012.1.13.3797921. PMID 23233555.
- Beals JK (November 19, 2008). "ASHG 2008: Gaucher Disease Mutation Carriers at Higher Risk for Parkinson's Disease". Medscape Medical News.
- Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R (November 2004). "Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews". The New England Journal of Medicine. 351 (19): 1972–7. doi:10.1056/NEJMoa033277. PMID 15525722.
- Arends M, van Dussen L, Biegstraaten M, Hollak CE (June 2013). "Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature". British Journal of Haematology. 161 (6): 832–42. doi:10.1111/bjh.12335. PMID 23594419.
- Thomas AS, Mehta A, Hughes DA (May 2014). "Gaucher disease: haematological presentations and complications". British Journal of Haematology. 165 (4): 427–40. doi:10.1111/bjh.12804. PMID 24588457.
- Ayto R, Hughes DA (2013). "Gaucher disease and myeloma". Critical Reviews in Oncogenesis. 18 (3): 247–68. doi:10.1615/critrevoncog.2013006061. PMID 23510067.
- Barth BM, Shanmugavelandy SS, Tacelosky DM, Kester M, Morad SA, Cabot MC (2013). "Gaucher's disease and cancer: a sphingolipid perspective". Critical Reviews in Oncogenesis. 18 (3): 221–34. doi:10.1615/critrevoncog.2013005814. PMC 3604879. PMID 23510065.
- "Gaucher Disease". symptoma. Retrieved 2015-12-07.
- Nagral A (March 2014). "Gaucher disease". Journal of Clinical and Experimental Hepatology. 4 (1): 37–50. doi:10.1016/j.jceh.2014.02.005. PMC 4017182. PMID 25755533.
- Bennett LL, Mohan D (September 2013). "Gaucher disease and its treatment options". The Annals of Pharmacotherapy. 47 (9): 1182–93. doi:10.1177/1060028013500469. PMID 24259734.
- Archived September 24, 2006, at the Wayback Machine
- Dreborg S, Erikson A, Hagberg B (March 1980). "Gaucher disease--Norrbottnian type. I. General clinical description". European Journal of Pediatrics. 133 (2): 107–18. doi:10.1007/BF00441578. PMID 7363908.
- Shemesh E, Deroma L, Bembi B, Deegan P, Hollak C, Weinreb NJ, Cox TM (March 2015). "Enzyme replacement and substrate reduction therapy for Gaucher disease". The Cochrane Database of Systematic Reviews (3): CD010324. doi:10.1002/14651858.CD010324.pub2. PMID 25812601.
- Deegan PB, Cox TM (2012). "Imiglucerase in the treatment of Gaucher disease: a history and perspective". Drug Design, Development and Therapy. 6: 81–106. doi:10.2147/DDDT.S14395. PMC 3340106. PMID 22563238.
- World Health Organization. Regulatory Matters WHO Drug Information 5:3 1991. p 123
- Aetna. Last reviewed 8 August 2014 Clinical Policy Bulletin Number: 0442: Enzyme-replacement Therapy for Lysosomal Storage Disorders
- FDA Prescription and Over-the-Counter Drug Product List. 32ND Edition Cumulative Supplement Number 3: March 2012. Additions/Deletions for Prescription Drug Product List
- "Shire Announces FDA Approval Of VPRIV(TM) (velaglucerase Alfa For Injection) For The Treatment Of Type I Gaucher Disease". Medicalnewstoday.com. Archived from the original on June 13, 2011. Retrieved 2012-08-13.
- Yukhananov A (1 May 2012). "U.S. FDA approves Pfizer/Protalix drug for Gaucher". Chicago Tribune. Reuters. Retrieved 2 May 2012.
- European Medicines Agency. Human Medicines Database. Zavesca (miglustat) Page Accessed 1 September 2014.
- European Medicines Agency 1 April 2003 Scientific discussion related to approval of Zavesca.
- "Center Watch: Cerdelga (eliglustat)".
- Gaucher Disease at National Gaucher Foundation. Retrieved June 2012
- "Gaucher Disease Genetics | About Gaucher Disease | National Gaucher Foundation". National Gaucher Foundation. Retrieved 2016-11-16.
- "Gaucher disease - Affected population". NORD - National Organization for Rare Disorders. Archived from the original on 25 September 2013. Retrieved 21 September 2013.
- Brady RO, Kanfer JN, Shapiro D (January 1965). "Metabolism of glucocerebrosides II. Evidence of an enzymatic deficiency in Gaucher's disease". Biochemical and Biophysical Research Communications. 18 (2): 221–5. doi:10.1016/0006-291X(65)90743-6. PMID 14282020.
- "Eating Fried Chicken - Cold is Gold". RNZ. 2019-08-01. Retrieved 2019-08-08.