Farber disease
Farber disease (also known as Farber's lipogranulomatosis, ceramidase deficiency, "Fibrocytic dysmucopolysaccharidosis," and "Lipogranulomatosis"[2]:545) is an extremely rare (80 cases reported worldwide to this day[3]) autosomal recessive lysosomal storage disease marked by a deficiency in the enzyme ceramidase that causes an accumulation of fatty material sphingolipids leading to abnormalities in the joints, liver, throat, tissues and central nervous system. Normally, the enzyme ceramidase breaks down fatty material in the body’s cells. In Farber disease, the gene responsible for making this enzyme is mutated. Hence, the fatty material is never broken down and, instead, accumulates in various parts of the body, leading to the signs and symptoms of this disorder.
| Farber disease | |
|---|---|
| Other names | Acid ceramidase deficiency[1] |
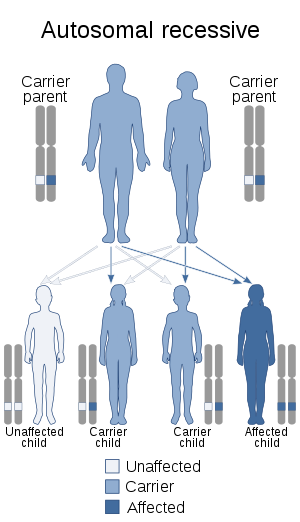 | |
| This condition is inherited in an autosomal recessive manner | |
| Specialty | Endocrinology |
Diagnosis
Disease onset is typically in early infancy but may occur later in life. Children who have the classic form of Farber disease develop symptoms within the first few weeks of life. These symptoms may include moderately impaired mental ability and problems with swallowing. The liver, heart and kidneys may also be affected. Other symptoms may include vomiting, arthritis, swollen lymph nodes, swollen joints, joint contractures (chronic shortening of muscles or tendons around joints), hoarseness and xanthomas which thicken around joints as the disease progresses. Patients with breathing difficulty may require a breathing tube.
Treatment
There is no specific treatment for Farber disease. Corticosteroids may be prescribed to relieve pain. Bone marrow transplants may improve granulomas (small masses of inflamed tissue) on patients with little or no lung or nervous system complications. Older patients may have granulomas surgically reduced or removed.
Prognosis
Most children with Farber disease die by age 2, usually from lung disease. In one of the most severe forms of the disease, an enlarged liver and spleen (hepatosplenomegaly) can be diagnosed soon after birth. Children born with this form of the disease usually die within 6 months.
Eponym
It is named for Sidney Farber.[5][6]
References
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Farber disease". www.orpha.net. Retrieved 17 April 2019.
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 978-0-7216-2921-6.
- "Farber lipogranulomatosis".
- Devi AR, Gopikrishna M, Ratheesh R, Savithri G, Swarnalata G, Bashyam M (2006). "Farber lipogranulomatosis: clinical and molecular genetic analysis reveals a novel mutation in an Indian family". J. Hum. Genet. 51 (9): 811–4. doi:10.1007/s10038-006-0019-z. PMID 16951918.
- synd/453 at Who Named It?
- Farber S (1952). "A lipid metabolic disorder: disseminated lipogranulomatosis; a syndrome with similarity to, and important difference from, Niemann-Pick and Hand-Schüller-Christian disease". American Journal of Diseases of Children. 84 (4): 499–500. PMID 12975849.
External links
| Classification | |
|---|---|
| External resources |