Functional genomics
Functional genomics is a field of molecular biology that attempts to describe gene (and protein) functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects (such as genome sequencing projects and RNA sequencing). Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional “gene-by-gene” approach.
Definition and goals of functional genomics
In order to understand functional genomics it is important to first define function. In their paper[1] Graur et al. define function in two possible ways. These are "Selected effect" and "Causal Role". The "Selected Effect" function refers to the function for which a trait(DNA, RNA, protein etc.) is selected for. The "Causal role" function refers to the function that a trait is sufficient and necessary for. Functional genomics usually tests the "Causal role" definition of function.
The goal of functional genomics is to understand the function of genes or proteins, eventually all components of a genome. The term functional genomics is often used to refer to the many technical approaches to study an organism's genes and proteins, including the "biochemical, cellular, and/or physiological properties of each and every gene product"[2] while some authors include the study of nongenic elements in their definition.[3] Functional genomics may also include studies of natural genetic variation over time (such as an organism's development) or space (such as its body regions), as well as functional disruptions such as mutations.
The promise of functional genomics is to generate and synthesize genomic and proteomic knowledge into an understanding of the dynamic properties of an organism. This could potentially provide a more complete picture of how the genome specifies function compared to studies of single genes. Integration of functional genomics data is often a part of systems biology approaches.
Techniques and applications
Functional genomics includes function-related aspects of the genome itself such as mutation and polymorphism (such as single nucleotide polymorphism (SNP) analysis), as well as the measurement of molecular activities. The latter comprise a number of "-omics" such as transcriptomics (gene expression), proteomics (protein production), and metabolomics. Functional genomics uses mostly multiplex techniques to measure the abundance of many or all gene products such as mRNAs or proteins within a biological sample. A more focused functional genomics approach might test the function of all variants of one gene and quantify the effects of mutants by using sequencing as a readout of activity. Together these measurement modalities endeavor to quantitate the various biological processes and improve our understanding of gene and protein functions and interactions.
At the DNA level
Genetic interaction mapping
Systematic pairwise deletion of genes or inhibition of gene expression can be used to identify genes with related function, even if they do not interact physically. Epistasis refers to the fact that effects for two different gene knockouts may not be additive; that is, the phenotype that results when two genes are inhibited may be different from the sum of the effects of single knockouts.
DNA/Protein interactions
Proteins formed by the translation of the mRNA (messenger RNA, a coded information from DNA for protein synthesis) play a major role in regulating gene expression. To understand how they regulate gene expression it is necessary to identify DNA sequences that they interact with. Techniques have been developed to identify sites of DNA-protein interactions. These include Chip-sequencing, CUT&RUN sequencing and Calling Cards.[4]
DNA accessibility assays
Assays have been developed to identify regions of the genome that are accessible. These regions of open chromatin are candidate regulatory regions. These assays include ATAC-seq, DNase-Seq and FAIRE-Seq.
At the RNA level
Microarrays
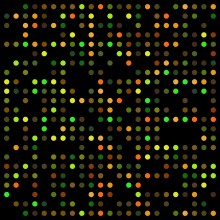
Microarrays measure the amount of mRNA in a sample that corresponds to a given gene or probe DNA sequence. Probe sequences are immobilized on a solid surface and allowed to hybridize with fluorescently labeled “target” mRNA. The intensity of fluorescence of a spot is proportional to the amount of target sequence that has hybridized to that spot, and therefore to the abundance of that mRNA sequence in the sample. Microarrays allow for identification of candidate genes involved in a given process based on variation between transcript levels for different conditions and shared expression patterns with genes of known function.
SAGE
Serial analysis of gene expression (SAGE) is an alternate method of analysis based on RNA sequencing rather than hybridization. SAGE relies on the sequencing of 10–17 base pair tags which are unique to each gene. These tags are produced from poly-A mRNA and ligated end-to-end before sequencing. SAGE gives an unbiased measurement of the number of transcripts per cell, since it does not depend on prior knowledge of what transcripts to study (as microarrays do).
RNA sequencing
RNA sequencing has taken over microarray and SAGE technology in recent years, as noted in 2016, and has become the most efficient way to study transcription and gene expression. This is typically done by next-generation sequencing.[5]
A subset of sequenced RNAs are small RNAs, a class of non-coding RNA molecules that are key regulators of transcriptional and post-transcriptional gene silencing, or RNA silencing. Next generation sequencing is the gold standard tool for non-coding RNA discovery, profiling and expression analysis.
Massively Parallel Reporter Assays (MPRAs)
Massively parallel reporter assays is a technology to test the cis-regulatory activity of DNA sequences.[6][7] MPRAs use a plasmid with a synthetic cis-regulatory element upstream of a promoter driving a synthetic gene such as Green Fluorescent Protein. A library of cis-regulatory elements is usually tested using MPRAs, a library can contain from hundreds to thousands of cis-regulatory elements. The cis-regulatory activity of the elements is assayed by using the downstream reporter activity. The activity of all the library members is assayed in parallel using barcodes for each cis-regulatory element. One limitation of MPRAs is that the activity is assayed on a plasmid and may not capture all aspects of gene regulation observed in the genome.
STARR-seq
STARR-seq is a technique similar to MPRAs to assay enhancer activity of randomly sheared genomic fragments. In the original publication,[8] randomly sheared fragments of the Drosophila genome were placed downstream of a minimal promoter. Candidate enhancers amongst the randomly sheared fragments will transcribe themselves using the minimal promoter. By using sequencing as a readout and controlling for input amounts of each sequence the strength of putative enhancers are assayed by this method.
Perturb-seq
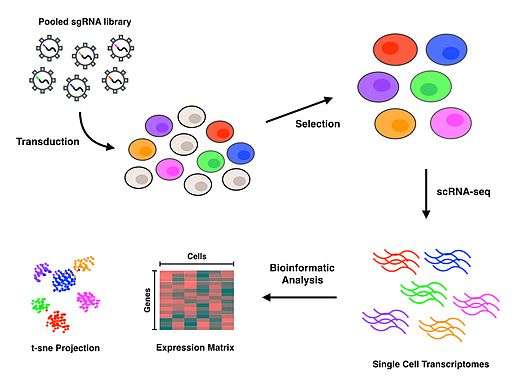
Perturb-seq couples CRISPR mediated gene knockdowns with single-cell gene expression. Linear models are used to calculate the effect of the knockdown of a single gene on the expression of multiple genes.
At the protein level
Yeast two-hybrid system
A yeast two-hybrid screening (Y2H) tests a "bait" protein against many potential interacting proteins ("prey") to identify physical protein–protein interactions. This system is based on a transcription factor, originally GAL4,[9] whose separate DNA-binding and transcription activation domains are both required in order for the protein to cause transcription of a reporter gene. In a Y2H screen, the "bait" protein is fused to the binding domain of GAL4, and a library of potential "prey" (interacting) proteins is recombinantly expressed in a vector with the activation domain. In vivo interaction of bait and prey proteins in a yeast cell brings the activation and binding domains of GAL4 close enough together to result in expression of a reporter gene. It is also possible to systematically test a library of bait proteins against a library of prey proteins to identify all possible interactions in a cell.
AP/MS
Affinity purification and mass spectrometry (AP/MS) is able to identify proteins that interact with one another in complexes. Complexes of proteins are allowed to form around a particular “bait” protein. The bait protein is identified using an antibody or a recombinant tag which allows it to be extracted along with any proteins that have formed a complex with it. The proteins are then digested into short peptide fragments and mass spectrometry is used to identify the proteins based on the mass-to-charge ratios of those fragments.
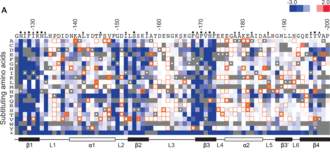
Deep Mutational Scanning
In Deep mutational scanning every possible amino acid change in a given protein is first synthesized. The activity of each of these protein variants is assayed in parallel using barcodes for each variant. By comparing the activity to the wild-type protein, the effect of each mutation is identified. While it is possible to assay every possible single amino-acid change due to combinatorics two or more concurrent mutations are hard to test. Deep Mutational scanning experiments have also been used to infer protein structure and protein-protein interactions.
Loss-of-function techniques
Mutagenesis
Gene function can be investigated by systematically “knocking out” genes one by one. This is done by either deletion or disruption of function (such as by insertional mutagenesis) and the resulting organisms are screened for phenotypes that provide clues to the function of the disrupted gene.
RNAi
RNA interference (RNAi) methods can be used to transiently silence or knock down gene expression using ~20 base-pair double-stranded RNA typically delivered by transfection of synthetic ~20-mer short-interfering RNA molecules (siRNAs) or by virally encoded short-hairpin RNAs (shRNAs). RNAi screens, typically performed in cell culture-based assays or experimental organisms (such as C. elegans) can be used to systematically disrupt nearly every gene in a genome or subsets of genes (sub-genomes); possible functions of disrupted genes can be assigned based on observed phenotypes.
CRISPR screens
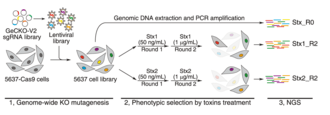
CRISPR-Cas9 has been used to delete genes in a multiplexed manner in cell-lines. Quantifying the amount of guide-RNAs for each gene before and after the experiment can point towards essential genes. If a guide-RNA disrupts an essential gene it will lead to the loss of that cell and hence there will be a depletion of that particular guide-RNA after the screen. In a recent CRISPR-cas9 experiment in mammalian cell-lines, around 2000 genes were found to be essential in multiple cell-lines.[11][12] Some of these genes were essential in only one cell-line. Most of genes are part of multi-protein complexes. This approach can be used to identify synthetic lethality by using the appropriate genetic background. CRISPRi and CRISPRa enable loss-of-function and gain-of-function screens in a similar manner. CRISPRi identified ~2100 essential genes in the K562 cell-line.[13][14] CRISPR deletion screens have also been used to identify potential regulatory elements of a gene. For example, a technique called ScanDel was published which attempted this approach. The authors deleted regions outside a gene of interest(HPRT1 involved in a Mendelian disorder) in an attempt to identify regulatory elements of this gene.[15] Gassperini et al. did not identify any distal regulatory elements for HPRT1 using this approach, however such approaches can be extended to other genes of interest.
Functional annotations for genes
Genome annotation
Putative genes can be identified by scanning a genome for regions likely to encode proteins, based on characteristics such as long open reading frames, transcriptional initiation sequences, and polyadenylation sites. A sequence identified as a putative gene must be confirmed by further evidence, such as similarity to cDNA or EST sequences from the same organism, similarity of the predicted protein sequence to known proteins, association with promoter sequences, or evidence that mutating the sequence produces an observable phenotype.
Rosetta stone approach
The Rosetta stone approach is a computational method for de-novo protein function prediction. It is based on the hypothesis that some proteins involved in a given physiological process may exist as two separate genes in one organism and as a single gene in another. Genomes are scanned for sequences that are independent in one organism and in a single open reading frame in another. If two genes have fused, it is predicted that they have similar biological functions that make such co-regulation advantageous.
Bioinformatics methods for Functional genomics
Because of the large quantity of data produced by these techniques and the desire to find biologically meaningful patterns, bioinformatics is crucial to analysis of functional genomics data. Examples of techniques in this class are data clustering or principal component analysis for unsupervised machine learning (class detection) as well as artificial neural networks or support vector machines for supervised machine learning (class prediction, classification). Functional enrichment analysis is used to determine the extent of over- or under-expression (positive- or negative- regulators in case of RNAi screens) of functional categories relative to a background sets. Gene ontology based enrichment analysis are provided by DAVID and gene set enrichment analysis (GSEA),[16] pathway based analysis by Ingenuity [17] and Pathway studio[18] and protein complex based analysis by COMPLEAT.[19]
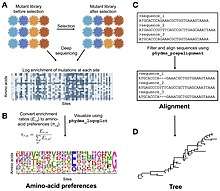
New computational methods have been developed for understanding the results of a deep mutational scanning experiment. 'phydms' compares the result of a deep mutational scanning experiment to a phylogenetic tree.[20] This allows the user to infer if the selection process in nature applies similar constraints on a protein as the results of the deep mutational scan indicate. This may allow an experimenter to choose between different experimental conditions based on how well they reflect nature. Deep mutational scanning has also been used to infer protein-protein interactions.[21] The authors used a thermodynamic model to predict the effects of mutations in different parts of a dimer. Deep mutational structure can also be used to infer protein structure. Strong positive epistasis between two mutations in a deep mutational scan can be indicative of two parts of the protein that are close to each other in 3-D space. This information can then be used to infer protein structure. A proof of principle of this approach was shown by two groups using the protein GB1.[22][23]
Results from MPRA experiments have required machine learning approaches to interpret the data. A gapped k-mer SVM model has been used to infer the kmers that are enriched within cis-regulatory sequences with high activity compared to sequences with lower activity.[24] These models provide high predictive power. Deep learning and random forest approaches have also been used to interpret the results of these high-dimensional experiments.[25] These models are beginning to help develop a better understanding of non-coding DNA function towards gene-regulation.
Consortium projects focused on Functional Genomics
The ENCODE project
The ENCODE (Encyclopedia of DNA elements) project is an in-depth analysis of the human genome whose goal is to identify all the functional elements of genomic DNA, in both coding and noncoding regions. Important results include evidence from genomic tiling arrays that most nucleotides are transcribed as coding transcripts, noncoding RNAs, or random transcripts, the discovery of additional transcriptional regulatory sites, further elucidation of chromatin-modifying mechanisms.
The Genotype-Tissue Expression (GTEx) project
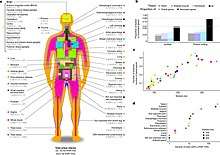
The GTEx project is a human genetics project aimed at understanding the role of genetic variation in shaping variation in the transcriptome across tissues. The project has collected a variety of tissue samples (> 50 different tissues) from more than 700 post-mortem donors. This has resulted in the collection of >11,000 samples. GTEx has helped understand the tissue-sharing and tissue-specificity of EQTLs.[26]
See also
- Systems biology
- Structural genomics
- Comparative genomics
- Pharmacogenomics
- MGED Society
- Epigenetics
- Bioinformatics
- Epistasis and functional genomics
- Synthetic viability
- Protein function prediction
References
- Graur D, Zheng Y, Price N, Azevedo RB, Zufall RA, Elhaik E (20 February 2013). "On the immortality of television sets: "function" in the human genome according to the evolution-free gospel of ENCODE". Genome Biology and Evolution. 5 (3): 578–90. doi:10.1093/gbe/evt028. PMC 3622293. PMID 23431001.
- Gibson G, Muse SV. A primer of genome science (3rd ed.). Sunderland, MA: Sinauer Associates.
- Pevsner J (2009). Bioinformatics and functional genomics (2nd ed.). Hoboken, NJ: Wiley-Blackwell.
- Wang H, Mayhew D, Chen X, Johnston M, Mitra RD (May 2011). "Calling Cards enable multiplexed identification of the genomic targets of DNA-binding proteins". Genome Research. 21 (5): 748–55. doi:10.1101/gr.114850.110. PMC 3083092. PMID 21471402.
- Hrdlickova R, Toloue M, Tian B (January 2017). "RNA-Seq methods for transcriptome analysis". Wiley Interdisciplinary Reviews: RNA. 8 (1): e1364. doi:10.1002/wrna.1364. PMC 5717752. PMID 27198714.
- Kwasnieski JC, Fiore C, Chaudhari HG, Cohen BA (October 2014). "High-throughput functional testing of ENCODE segmentation predictions". Genome Research. 24 (10): 1595–602. doi:10.1101/gr.173518.114. PMC 4199366. PMID 25035418.
- Patwardhan RP, Hiatt JB, Witten DM, Kim MJ, Smith RP, May D, et al. (February 2012). "Massively parallel functional dissection of mammalian enhancers in vivo". Nature Biotechnology. 30 (3): 265–70. doi:10.1038/nbt.2136. PMC 3402344. PMID 22371081.
- Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, Stark A (March 2013). "Genome-wide quantitative enhancer activity maps identified by STARR-seq". Science. 339 (6123): 1074–7. Bibcode:2013Sci...339.1074A. doi:10.1126/science.1232542. PMID 23328393.
- Fields S, Song O (July 1989). "A novel genetic system to detect protein-protein interactions". Nature. 340 (6230): 245–6. Bibcode:1989Natur.340..245F. doi:10.1038/340245a0. PMID 2547163.
- Tian S, Muneeruddin K, Choi MY, Tao L, Bhuiyan RH, Ohmi Y, Furukawa K, Furukawa K, Boland S, Shaffer SA, Adam RM, Dong M (27 November 2018). "Genome-wide CRISPR screens for Shiga toxins and ricin reveal Golgi proteins critical for glycosylation". PLOS Biology. 16 (11). e2006951. doi:10.1371/journal.pbio.2006951. PMC 6258472. PMID 30481169.
- Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, et al. (December 2015). "High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities". Cell. 163 (6): 1515–26. doi:10.1016/j.cell.2015.11.015. PMID 26627737.
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. (January 2014). "Genome-scale CRISPR-Cas9 knockout screening in human cells". Science. 343 (6166): 84–87. Bibcode:2014Sci...343...84S. doi:10.1126/science.1247005. PMC 4089965. PMID 24336571.
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, et al. (October 2014). "Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation". Cell. 159 (3): 647–61. doi:10.1016/j.cell.2014.09.029. PMC 4253859. PMID 25307932.
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, et al. (September 2016). "Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation". eLife. 5. doi:10.7554/eLife.19760. PMC 5094855. PMID 27661255.
- Gasperini, Molly; Findlay, Gregory M.; McKenna, Aaron; Milbank, Jennifer H.; Lee, Choli; Zhang, Melissa D.; Cusanovich, Darren A.; Shendure, Jay (August 2017). "CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions". The American Journal of Human Genetics. 101 (2): 192–205. doi:10.1016/j.ajhg.2017.06.010. PMC 5544381. PMID 28712454.
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. (October 2005). "Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles". Proceedings of the National Academy of Sciences of the United States of America. 102 (43): 15545–50. Bibcode:2005PNAS..10215545S. doi:10.1073/pnas.0506580102. PMC 1239896. PMID 16199517.
- "Ingenuity Systems". Archived from the original on 1999-01-25. Retrieved 2007-12-31.
- "Ariadne Genomics: Pathway Studio". Archived from the original on 2007-12-30. Retrieved 2007-12-31.
- Vinayagam A, Hu Y, Kulkarni M, Roesel C, Sopko R, Mohr SE, Perrimon N (February 2013). "Protein complex-based analysis framework for high-throughput data sets". Science Signaling. 6 (264): rs5. doi:10.1126/scisignal.2003629. PMC 3756668. PMID 23443684.
- Hilton SK, Doud MB, Bloom JD (2017). "phydms: software for phylogenetic analyses informed by deep mutational scanning". PeerJ. 5: e3657. doi:10.7717/peerj.3657. PMC 5541924. PMID 28785526.
- Diss G, Lehner B (April 2018). "The genetic landscape of a physical interaction". eLife. 7. doi:10.7554/eLife.32472. PMC 5896888. PMID 29638215.
- Schmiedel, Jörn M.; Lehner, Ben (17 June 2019). "Determining protein structures using deep mutagenesis". Nature Genetics. 51 (7): 1177–1186. doi:10.1038/s41588-019-0431-x. PMID 31209395.
- Rollins, Nathan J.; Brock, Kelly P.; Poelwijk, Frank J.; Stiffler, Michael A.; Gauthier, Nicholas P.; Sander, Chris; Marks, Debora S. (17 June 2019). "Inferring protein 3D structure from deep mutation scans". Nature Genetics. 51 (7): 1170–1176. doi:10.1038/s41588-019-0432-9. PMC 7295002. PMID 31209393.
- Ghandi M, Lee D, Mohammad-Noori M, Beer MA (July 2014). "Enhanced regulatory sequence prediction using gapped k-mer features". PLOS Computational Biology. 10 (7): e1003711. Bibcode:2014PLSCB..10E3711G. doi:10.1371/journal.pcbi.1003711. PMC 4102394. PMID 25033408.
- Li Y, Shi W, Wasserman WW (May 2018). "Genome-wide prediction of cis-regulatory regions using supervised deep learning methods". BMC Bioinformatics. 19 (1): 202. doi:10.1186/s12859-018-2187-1. PMC 5984344. PMID 29855387.
- GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (Ldacc)—Analysis Working Group.; Statistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; Biospecimen Collection Source Site—RPCI; Biospecimen Core Resource—VARI; Brain Bank Repository—University of Miami Brain Endowment Bank; Leidos Biomedical—Project Management; ELSI Study; Genome Browser Data Integration &Visualization—EBI; Genome Browser Data Integration &Visualization—Ucsc Genomics Institute, University of California Santa Cruz.; Lead analysts; Laboratory, Data Analysis &Coordinating Center (Ldacc):.; NIH program management; Biospecimen collection; Pathology; eQTL manuscript working group; Battle, A.; Brown, C. D.; Engelhardt, B. E.; Montgomery, S. B. (12 October 2017). "Genetic effects on gene expression across human tissues" (PDF). Nature. 550 (7675): 204–213. Bibcode:2017Natur.550..204A. doi:10.1038/nature24277. PMC 5776756. PMID 29022597.
External links
- Functional genomics: An introduction to the EBI resources on Train OnLine
- European Science Foundation Programme on Frontiers of Functional Genomics
- MUGEN NoE — Integrated Functional Genomics in Mutant Mouse Models
- Nature insights: functional genomics
- Bioinformatics and functional genomics — Companion site for Bioinformatics and functional genomics, 2nd ed.
- ENCODE
- 4th European Science Foundation Conference on Functional Genomics and Disease
| Genomics | |
|---|---|
| Bioinformatics | |
| Structural biology |
|
| Research tools |
|
| Organizations |
|
| |