Perturb-seq
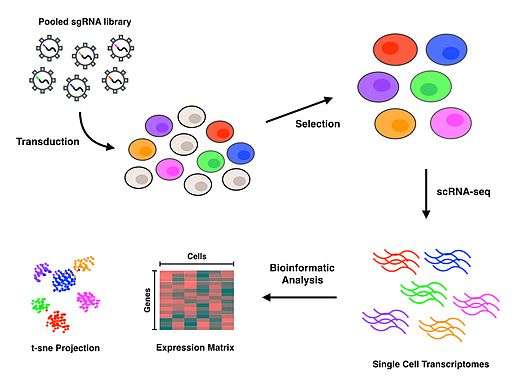
Perturb-seq (also known as CRISP-seq and CROP-seq) refers to a high-throughput method of performing single cell RNA sequencing (scRNA-seq) on pooled genetic perturbation screens.[1][2][3] Perturb-seq combines multiplexed CRISPR mediated gene inactivations with single cell RNA sequencing to assess comprehensive gene expression phenotypes for each perturbation. Inferring a gene’s function by applying genetic perturbations to knock down or knock out a gene and studying the resulting phenotype is known as reverse genetics. Perturb-seq is a reverse genetics approach that allows for the investigation of phenotypes at the level of the transcriptome, to elucidate gene functions in many cells, in a massively parallel fashion.
The Perturb-seq protocol uses CRISPR technology to inactivate specific genes and DNA barcoding of each guide RNA to allow for all perturbations to be pooled together and later deconvoluted, with assignment of each phenotype to a specific guide RNA.[1][2] Droplet-based microfluidics platforms (or other cell sorting and separating techniques) are used to isolate individual cells, and then scRNA-seq is performed to generate gene expression profiles for each cell. Upon completion of the protocol, bioinformatics analyses are conducted to associate each specific cell and perturbation with a transcriptomic profile that characterizes the consequences of inactivating each gene.
In the December 2016 issue of the Cell journal, two companion papers were published that each introduced and described this technique.[1][2] A third paper describing a conceptually similar approach (termed CRISP-seq) was also published in the same issue.[4] In October 2016, the CROP-seq method for single-cell CRISPR screening was presented in a preprint on bioRxiv[5] and later published in the Nature Methods journal.[3] While each paper shared the core principles of combining CRISPR mediated perturbation with scRNA-seq, their experimental, technological and analytical approaches differed in several aspects, to explore distinct biological questions, demonstrating the broad utility of this methodology. For example, the CRISPR-seq paper demonstrated the feasibility of in vivo studies using this technology, and the CROP-seq protocol facilitates large screens by providing a vector that makes the guide RNA itself readable (rather than relying on expressed barcodes), which allows for single-step guide RNA cloning.[6]
Experimental workflow
CRISPR Single Guide RNA Library design and selection
Pooled CRISPR libraries that enable gene inactivation can come in the form of either knockout or interference. Knockout libraries perturb genes through double stranded breaks that prompt the error prone non-homologous end joining repair pathway to introduce disruptive insertions or deletions. CRISPR interference (CRISPRi) on the other hand utilizes a catalytically inactive nuclease to physically block RNA polymerase, effectively preventing or halting transcription.[7] Perturb-seq has been utilized with both the knockout and CRISPRi approaches in the Dixit et al. paper [2] and the Adamson et al. paper,[1] respectively.
Pooling all guide RNAs into a single screen relies on DNA barcodes that act as identifiers for each unique guide RNA. There are several commercially available pooled CRISPR libraries including the guide barcode library used in the study by Adamson et al.[1] CRISPR libraries can also be custom made using tools for sgRNA design, many of which are listed on the CRISPR/cas9 tools Wikipedia page.
Lentiviral vectors
The sgRNA expression vector design will depend largely on the experiment performed but requires the following central components:
- Promoter
- Restriction sites
- Primer Binding Sites
- sgRNA
- Guide Barcode
- Reporter gene:
- Fluorescent gene: vectors are often constructed to include a gene encoding a fluorescent protein, such that successfully transduced cells can be visually and quantitatively assessed by their expression.
- Antibiotic resistance gene: similar to fluorescent markers, antibiotic resistance genes are often incorporated into vectors to allow for selection of successfully transduced cells.
- CRISPR-associated endonuclease: Cas9 or other CRISPR-associated endonucleases such as Cpf1 must be introduced to cells that do not endogenously express them. Due to the large size of these genes, a two-vector system can be used to express the endonuclease separately from the sgRNA expression vector.[8]
Transduction and selection
Cells are typically transduced with a Multiplicity of Infection (MOI) of 0.4 to 0.6 lentiviral particles per cell to maximize the likelihood of obtaining the most amount of cells which contain a single guide RNA.[8][9] If the effects of simultaneous perturbations are of interest, a higher MOI may be applied to increase the amount of transduced cells with more than one guide RNA. Selection for successfully transduced cells is then performed using a fluorescence assay or an antibiotic assay, depending on the reporter gene used in the expression vector.
Single-cell library preparation
After successfully transduced cells have been selected for, isolation of single cells is needed to conduct scRNA-seq. Perturb-seq and CROP-seq have been performed using droplet-based technology for single cell isolation,[1][2][3] while the closely related CRISP-seq was performed with a microwell-based approach.[4] Once cells have been isolated at the single cell level, reverse transcription, amplification and sequencing takes place to produce gene expression profiles for each cell. Many scRNA-seq approaches incorporate unique molecular identifiers (UMIs) and cell barcodes during the reverse transcription step to index individual RNA molecules and cells, respectively. These additional barcodes serve to help quantify RNA transcripts and to associate each of the sequences with their cell of origin.
Bioinformatics analysis
Read alignment and processing are performed to map quality reads to a reference genome. Deconvolution of cell barcodes, guide barcodes and UMIs enables the association of guide RNAs with the cells that contain them, thus allowing the gene expression profile of each cell to be affiliated with a particular perturbation. Further downstream analyses on the transcriptional profiles will depend entirely on the biological question of interest. T-distributed Stochastic Neighbor Embedding (t-SNE) is a commonly used machine learning algorithm to visualize the high-dimensional data that results from scRNA-seq in a 2-dimensional scatterplot.[1][4][10] The authors who first performed Perturb-seq developed an in-house computational framework called MIMOSCA that predicts the effects of each perturbation using a linear model and is available on an open software repository.[11]
Advantages and limitations
Perturb-seq makes use of current technologies in molecular biology to integrate a multi-step workflow that couples high-throughput screening with complex phenotypic outputs. When compared to alternative methods used for gene knockdowns or knockouts, such as RNAi, zinc finger nucleases or transcription activator-like effector nucleases (TALENs), the application of CRISPR-based perturbations enables more specificity, efficiency and ease of use.[8][12] Another advantage of this protocol is that while most screening approaches can only assay for simple phenotypes, such as cellular viability, scRNA-seq allows for a much richer phenotypic readout, with quantitative measurements of gene expression in many cells simultaneously.
However, while a large and comprehensive amount of data can be a benefit, it can also present a major challenge. Single cell RNA expression readouts are known to produce ‘noisy’ data, with a significant number of false positives.[13] Both the large size and noise that is associated with scRNA-seq will likely require new and powerful computational methods and bioinformatics pipelines to better make sense of the resulting data. Another challenge associated with this protocol is the creation of large scale CRISPR libraries. The preparation of these extensive libraries depends upon a comparative increase in the resources required to culture the massive numbers of cells that are needed to achieve a successful screen of many perturbations.[8]
In parallel to these single-cell methods, other approaches have been developed to reconstruct genetic pathways using whole-organism RNA-sequencing. These methods use a single aggregate statistic, called the transcriptome-wide epistasis coefficient, to guide pathway reconstruction.[14] In contrast with the statistical framework of the methods described above, this coefficient may be more robust to noise and is intuitively interpretable in terms of Batesonian epistasis. This approach was used to identify a new state in the life cycle of the nematode C. elegans.[15]
Applications
Perturb-seq or other conceptually similar protocols can be used to address a broad scope of biological questions and the applications of this technology will likely grow over time. Three papers on this topic, published in the December 2016 issue of the Journal Cell, demonstrated the utility of this method by applying it to the investigation of several distinct biological functions. In the paper, “Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens”, the authors used Perturb-seq to conduct knockouts of transcription factors related to the immune response in hundreds of thousands of cells to investigate the cellular consequences of their inactivation.[2] They also explored the effects of transcription factors on cell states in the context of the cell cycle. In the study led by UCSF, “A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response” the researchers suppressed multiple genes in each cell to study the unfolded protein response (UPR) pathway.[1] With a similar methodology, but using the term CRISP-seq instead of Perturb-seq, the paper "Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq" performed a proof of concept experiment by using the technique to probe regulatory pathways related to innate immunity in mice.[4] Lethality of each perturbation and epistasis analyses in cells with multiple perturbations was also investigated in these papers. Perturb-seq has so far been used with very few perturbations per experiment, but it can theoretically be scaled up to address the whole genome. Finally, the October 2016 preprint[5] and subsequent paper[3] demonstrate the bioinformatic reconstruction of the T cell receptor signaling pathway in Jurkat cells based on CROP-seq data.
While these publications used these protocols for answering complex biological questions, this technology can also be used as a validation assay to ensure the specificity of any CRISPR based knockdown or knockout; the expression levels of the target genes as well as others can be measured with single cell resolution in parallel, to detect whether the perturbation was successful and to assess the experiment for off target effects. Furthermore, these protocols make it possible to perform perturbation screens in heterogeneous tissues, while obtaining cell type specific gene expression responses.
References
- Adamson, Britt; Norman, Thomas M.; Jost, Marco; Cho, Min Y.; Nuñez, James K.; Chen, Yuwen; Villalta, Jacqueline E.; Gilbert, Luke A.; Horlbeck, Max A. (2016). "A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response". Cell. 167 (7): 1867–1882.e21. doi:10.1016/j.cell.2016.11.048. PMC 5315571. PMID 27984733.
- Dixit, Atray; Parnas, Oren; Li, Biyu; Chen, Jenny; Fulco, Charles P.; Jerby-Arnon, Livnat; Marjanovic, Nemanja D.; Dionne, Danielle; Burks, Tyler (2016). "Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens". Cell. 167 (7): 1853–1866.e17. doi:10.1016/j.cell.2016.11.038. PMC 5181115. PMID 27984732.
- Datlinger, Paul; Rendeiro, André F; Schmidl, Christian; Krausgruber, Thomas; Traxler, Peter; Klughammer, Johanna; Schuster, Linda C; Kuchler, Amelie; Alpar, Donat (2017). "Pooled CRISPR screening with single-cell transcriptome readout". Nature Methods. 14 (3): 297–301. doi:10.1038/nmeth.4177. PMC 5334791. PMID 28099430.
- Jaitin, Diego Adhemar; Weiner, Assaf; Yofe, Ido; Lara-Astiaso, David; Keren-Shaul, Hadas; David, Eyal; Salame, Tomer Meir; Tanay, Amos; Oudenaarden, Alexander van (2016). "Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq". Cell. 167 (7): 1883–1896.e15. doi:10.1016/j.cell.2016.11.039. PMID 27984734.
- Datlinger, Paul; Schmidl, Christian; Rendeiro, Andre F.; Traxler, Peter; Klughammer, Johanna; Schuster, Linda; Bock, Christoph (2016-10-27). "Pooled CRISPR screening with single-cell transcriptome read-out". bioRxiv 10.1101/083774.
- "Pooled CRISPR screening with single-cell transcriptome readout". crop-seq.computational-epigenetics.org. Retrieved 2017-05-30.
- Larson, Matthew H; Gilbert, Luke A; Wang, Xiaowo; Lim, Wendell A; Weissman, Jonathan S; Qi, Lei S (2013). "CRISPR interference (CRISPRi) for sequence-specific control of gene expression". Nature Protocols. 8 (11): 2180–2196. doi:10.1038/nprot.2013.132. PMC 3922765. PMID 24136345.
- Shalem, Ophir; Sanjana, Neville E.; Hartenian, Ella; Shi, Xi; Scott, David A.; Mikkelsen, Tarjei S.; Heckl, Dirk; Ebert, Benjamin L.; Root, David E. (2014-01-03). "Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells". Science. 343 (6166): 84–87. doi:10.1126/science.1247005. hdl:1721.1/111576. ISSN 0036-8075. PMC 4089965. PMID 24336571.
- Wang, Tim; Wei, Jenny J.; Sabatini, David M.; Lander, Eric S. (2014-01-03). "Genetic Screens in Human Cells Using the CRISPR-Cas9 System". Science. 343 (6166): 80–84. doi:10.1126/science.1246981. ISSN 0036-8075. PMC 3972032. PMID 24336569.
- Wilson, Nicola K.; Kent, David G.; Buettner, Florian; Shehata, Mona; Macaulay, Iain C.; Calero-Nieto, Fernando J.; Castillo, Manuel Sánchez; Oedekoven, Caroline A.; Diamanti, Evangelia (2015). "Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations". Cell Stem Cell. 16 (6): 712–724. doi:10.1016/j.stem.2015.04.004. PMC 4460190. PMID 26004780.
- https://github.com/asncd/MIMOSCA
- Boettcher, Michael; McManus, Michael T. (2015). "Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR". Molecular Cell. 58 (4): 575–585. doi:10.1016/j.molcel.2015.04.028. PMC 4441801. PMID 26000843.
- Liu, Serena; Trapnell, Cole (2016-02-17). "Single-cell transcriptome sequencing: recent advances and remaining challenges". F1000Research. 5: 182. doi:10.12688/f1000research.7223.1. PMC 4758375. PMID 26949524.
- Angeles-Albores, David; Puckett Robinson, Carmie; Williams, Brian A; Wold, Barbara J.; Sternberg, Paul W. (2018-03-27). "Reconstructing a metazoan genetic pathway with transcriptome-wide epistasis measurements". PNAS. 115 (13): E2930–E2939. doi:10.1073/pnas.1712387115. PMC 5879656. PMID 29531064.
- Angeles-Albores, David; Leighton, Daniel H.W.; Tsou, Tiffany; Khaw, Tiffany H.; Antoshechkin, Igor; Sternberg, Paul W. (2017-09-07). "The Caenorhabditis elegans Female-Like State: Decoupling the Transcriptomic Effects of Aging and Sperm Status". G3: Genes, Genomes, Genetics. 115 (9): 2969–2977. doi:10.1534/g3.117.300080. PMC 5592924. PMID 28751504.