Bacteriophage T12
Bacteriophage T12 is a bacteriophage that infects the bacterial species Streptococcus pyogenes, and converts a harmless strain of bacteria into a virulent strain. It carries the speA gene which codes for erythrogenic toxin A.[1] speA is also known as streptococcal pyogenic exotoxin A, scarlet fever toxin A, or even scarlatinal toxin.[2][3] Note that the name of the gene "speA" is italicized; the name of the toxin "speA" is not italicized. Erythrogenic toxin A converts a harmless, non-virulent strain of Streptococcus pyogenes to a virulent strain through lysogeny, a life cycle which is characterized by the ability of the genome to become a part of the host cell and be stably maintained there for generations.[4] Phages with a lysogenic life cycle are also called temperate phages.[1] Bacteriophage T12, a member of a family of related speA-carrying bacteriophages, is also a prototypic phage for all the speA-carrying phages of Streptococcus pyogenes, meaning that its genome is the prototype for the genomes of all such phages of S. pyogenes.[5] It is the main suspect as the cause of scarlet fever, an infectious disease that affects small children.[4]
| Bacteriophage T12 | |
|---|---|
| Virus classification | |
| Group: | Group I (dsDNA) |
| Order: | Unclassified |
Discovery and further research
The possibility of bacteriophage involvement in speA production was first introduced in 1926 when Cantacuzene and Boncieu reported that nonvirulent strains of S.pyogenes were transformed to virulent strains through some transferable element. Frobisher and Brown reported similar results in 1927, and in 1949, the reports were confirmed by Bingel [6][7] Later, in 1964, Zabriskie reported that phage T12 could cause speA production by lysogeny in strains that it became a part of.[8] In 1980, Johnson, Schlievert and Watson were able to confirm this and show that the gene for speA production was transferred from toxigenic strains of bacteria to non-toxigenic strains through lysogeny. In their experiment, every transformed, toxin-producing bacterial colony was lysogenic, i.e. contained the T12 gene. In addition, none of the colonies containing the T12 genome was negative for speA, and therefore, the conclusion was drawn that all lysogens produced the toxin.[9] However, McKane and Ferretti reported, in 1981, that a mutant of phage T12 induced speA production virulently. This mutant, the bacteriophage T253, entered the lytic cycle, a life cycle in which the host cell is destroyed.[10] In 1983, Johnson and Schlievert published a map of the T12 genome, revealing also that three rounds of packaging occur in the genome.[8] The very next year, Johnson and Schlievert and Weeks and Ferreti also found, independently, that the bacteriophage T12 carries the structural gene for speA.[7][11] In 1986, Johson, Tomai and Schlievert mapped the attachment site (attP) for T12 adjacent to the speA gene, and established that all bacterial strains producing the toxin carry either phage T12 itself, or a closely related bacteriophage.[4] And finally, in 1997, McShan and Ferretti published that they had found the second attachment site (attR) for T12, while also revealing in another publication, which was also credited to Tang, that bacteriophage T12 inserts into a gene that encodes a serine tRNA in the host.[1][5]
Genome

The physical map of the T12 genome was found to be circular with a total length of 36.0kb.[8] The phage genome is reported to carry the speA gene,[11] which is a 1.7kb segment of the phage T12 genome flanked by SalI and HindIII sites.[7]
The phage integrase gene (int) and the phage attachment site (attp) are located just upstream of the speA gene in the phage genome. The bacteriophage T12 integrates into S. pyogenes chromosome by site-specific recombination into the anticodon loop of a gene that codes for serine tRNA. The bacterial attachment site (attB) has a 96 base pair sequence homologous to the phage attachment site and is located at the 3’ end of the tRNA gene such that the coding sequence of the tRNA gene remains intact after integration of the prophage. Phage T12 is the first example of a phage from a gram-positive, low G-C content host that uses this kind of integration site.[1][5]
Role in pathogenesis
Diseases like scarlet fever and Streptococcal toxic shock syndrome are caused by lysogenized streptococcal strains that produce speA. The diseases are systemic responses to the speA circulating within the body.[12]
Scarlet fever
Scarlet fever, also known as scarletina, is so called because of the characteristic bright red rash it causes. It is most common in children between four and eight years of age.[13]
Signs and symptoms
The first stage of scarlet fever is typically strep throat (streptococcal pharyngitis) characterized by sore throat, fever, headache and sometimes nausea and vomiting. In two to three days, this is followed by the appearance of a diffuse erythematous rash that has a sandpaper texture. The rash first appears on the neck, then spreads to the chest, back and body extremities. A yellowish white coating covers the tongue, and is later shed, leaving the tongue with a strawberry appearance and swollen papillae. The rash fades away after five to six days of the onset of the disease, and is followed by peeling of skin, particularly over the hands and feet.[13][14][15]
Treatment
Penicillin, an antibiotic, is the drug of choice for the treatment of scarlet fever as for any other S. pyogenes infection. For those who are allergic to penicillin, the antibiotics erythromycin or clindamycin can be used. However, occasional resistance to these drugs has been reported.[16]
Streptococcal toxic shock syndrome
In streptococcal toxic shock syndrome (StrepTSS), speA produced by infected streptococcal strains acts as a superantigen and interacts with human monocytes and T lymphocytes, inducing T-cell proliferation and production of monokines (e.g. tumor necrosis factor α, interleukin 1, interleukin 6), and lymphokines (e.g. tumor necrosis factor β, interleukin 2, and gamma-interferon). These cytokines(TNFα, TNFβ) seem to mediate the fever, shock and organ failure characteristic of the disease.[12][17][18]
Signs and symptoms
Strep TSS is an acute, febrile illness that begins with a mild viral-like syndrome characterized by fever, chills, myalgia, diarrhea, vomiting and nausea and involves minor soft-tissue infection that may progress to shock, multi-organ failure, and death.[18]
Treatment
While penicillin is an effective treatment of mild infection, it is less effective in a severe case. Emerging treatments for strep TSS include clindamycin and intravenous gamma-globulin.[18]
Detection and elimination
The presence of lysogenic bacteriophage T12 can be tested through plaque assays if the indicator strain utilized is susceptible to the phage being tested. Plaque assays consist of pouring a soft agar solution with an indicator strain onto an agar plate. The indicator strain should be a strain of bacteria that can be infected by the phage that needs to be detected. After the soft agar is set the samples that are being tested for phage presence are then spread-plated onto the soft agar plates. The plates are then incubated overnight and checked for clearings (plaques) the next day. If the phage is present, indicator strains will become infected and go through the normal lysogenic cycle while the plates incubate, and then undergo lysis. The plaque that determines whether the phage is present or not is caused by the lysis of the indicator strains. Titers of plaques can be found by diluting the samples and counting plaque-forming units (PFUs).[19]
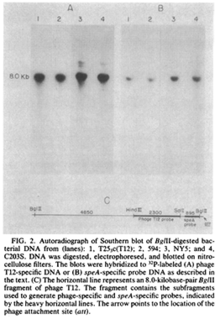
Biochemical tests such as Southern blots can also be used to detect the speA that the phage produces from the speA gene. This was done in research by Johnson, Tomai and Schlievert in 1985 by isolating the DNA of Streptococcal strains and running a restriction digest using BglII. After the digest was complete, the DNA samples were run on gel to separate the DNA. The DNA from this gel was then transferred to nitrocellulose paper and incubated with probes specific for speA. An image of this Southern blot can be seen in this article.[4]
Bacteriophages are very easily spread.[20] At lower exposures, Ultraviolet light can enhance the production of both phage T12 and speA.[3] Longer UV exposure times can kill the phage. UV light stresses lysogenic bacteria, leading the phages to propagate and burst the host bacterial cells.[21] In the case of T12, exposure to UV light increases the propagation of bacteriophage T12 at 20 seconds of exposure. After 20 seconds of exposure the UV light starts to kill the bacteriophage by damaging its genome.[22]
References
- W. M. McShan, Y. F. Tang, J. J. Ferretti (1997). "Bacteriophage T12 of Streptococcus pyogenes integrates into the gene encoding a serine tRNA". Molecular Microbiology. 23 (4): 719–28. doi:10.1046/j.1365-2958.1997.2591616.x. PMID 9157243.
- Stevens, Dennis L.; Tanner, Martha H.; Winship, Jay; Swarts, Raymond; Ries, Kristen M.; Patrick M. Schlievert; Edward Kaplan (6 July 1989). "Severe Group A Streptococcal Infections Associated with a Toxic Shock-like Syndrome and Scarlet Fever Toxin A". New England Journal of Medicine. 321 (1): 1–7. doi:10.1056/NEJM198907063210101. PMID 2659990.
- P. L. Wagner, M. K. Waldor (2002). "Bacteriophage control of bacterial virulence". Infection and Immunity. 70 (8): 3985–93. doi:10.1128/IAI.70.8.3985-3993.2002. PMC 128183. PMID 12117903.
- L. P. Johnson, M. A. Tomai, P. M. Schlievert (1986). "Bacteriophage involvement in group a streptococcal pyogenic exotoxin a production". Journal of Bacteriology. 166 (2): 623–7. doi:10.1128/jb.166.2.623-627.1986. PMC 214650. PMID 3009415.
- W. M. McShan; J. J. Ferretti (October 1997). "Genetic diversity in temperate bacteriophages of Streptococcus pyogenes: identification of a second attachment site for phages carrying the erythrogenic toxin A gene". Journal of Bacteriology. 179 (20): 6509–11. doi:10.1128/jb.179.20.6509-6511.1997. PMC 179571. PMID 9335304.
- Ferretti, Joseph; S. Kay Nida (May 1982). "Phage Influence on the Synthesis of Extracellular Toxins in Group A Streptococci". Infection and Immunity. 36 (2): 745–750. PMC 351293. PMID 7044976.
- Weeks CR, Ferretti JJ (1984). "The gene for type a streptococcal exotoxin (erythrogenic toxin) is located in bacteriophage T12". Infection and Immunity. 46 (2): 531–6. PMC 261567. PMID 6389348.
- Johnson, LP; Schlievert, PM (1983). "A physical map of the group A streptococcal pyrogenic exotoxin bacteriophage T12 genome". Molecular & General Genetics : MGG. 189 (2): 251–5. doi:10.1007/BF00337813. PMID 6304466.
- Johnson, LP; Schlievert, PM; D. W. Watson (April 1980). "Transfer of group A streptococcal pyrogenic exotoxin production to nontoxigenic strains of lysogenic conversion". Infection and Immunity. 28 (1): 254–7. PMC 550920. PMID 6991440.
- L. McKane; J. J. Ferretti (December 1981). "Phage-host interactions and the production of type A streptococcal exotoxin in group A streptococci". Infection and Immunity. 34 (3): 915–9. PMC 350956. PMID 7037644.
- Johnson LP, Schlievert PM (1984). "Group a streptococcal phage T12 carries the structural gene for pyrogenic exotoxin type A". Molecular & General Genetics. 194 (1–2): 52–6. doi:10.1007/BF00383496. PMID 6374381.
- Musser, JM; Hauser, AR; Kim, MH; Schlievert, PM; Nelson, K; Selander, RK (Apr 1, 1991). "Streptococcus pyogenes causing toxic-shock-like syndrome and other invasive diseases: clonal diversity and pyrogenic exotoxin expression". Proceedings of the National Academy of Sciences of the United States of America. 88 (7): 2668–72. Bibcode:1991PNAS...88.2668M. doi:10.1073/pnas.88.7.2668. PMC 51299. PMID 1672766.
- "Scarlet fever".
- Dennis L. Stevens. "Streptococcus pyogenes (Group A β-hemolytic Streptococcus)". Archived from the original on 2012-12-15.
- "Streptococcal Infections (S. pyogenes — Group A streptococci)".
- "Streptococcal Infections (Invasive Group A Strep)". Archived from the original on 2012-11-06.
- Hackett, SP; Stevens, DL (May 1992). "Streptococcal toxic shock syndrome: synthesis of tumor necrosis factor and interleukin-1 by monocytes stimulated with pyrogenic exotoxin A and streptolysin O.". The Journal of Infectious Diseases. 165 (5): 879–85. CiteSeerX 10.1.1.547.813. doi:10.1093/infdis/165.5.879. PMID 1569337.
- Dennis L. Stevens (2000). "Streptococcal toxic shock syndrome associated with necrotizing fasciitis". Annual Review of Medicine. 51: 271–88. doi:10.1146/annurev.med.51.1.271. PMID 10774464.
- Marie Panec. "Plaque Assay Protocols". Microbe Library. American Society for Microbiology. Archived from the original on 30 November 2012. Retrieved 28 November 2012.
- Ramirez E, Carbonell X, Villaverde A (2001). "Phage spread dynamics in clonal bacterial populations is depending on features of the founder cell". Microbiological Research. 156 (1): 35–40. doi:10.1078/0944-5013-00087. PMID 11372651.
- S. Atsumi, J. W. Little (2006). "Role of the lytic repressor in prophage induction of phage lambda as analyzed by a module-replacement approach". Proceedings of the National Academy of Sciences of the United States of America. 103 (12): 4558–63. Bibcode:2006PNAS..103.4558A. doi:10.1073/pnas.0511117103. PMC 1450210. PMID 16537413.
- Zabriskie JB (1964). "The Role of Temperate Bacteriophage in the Production of Erythrogenic Toxin by Group a Streptococci". The Journal of Experimental Medicine. 119 (5): 761–80. doi:10.1084/jem.119.5.761. PMC 2137738. PMID 14157029.