Pi backbonding
π backbonding, also called π backdonation, is a concept from chemistry in which electrons move from an atomic orbital on one atom to an appropriate symmetry antibonding orbital on a π-acceptor ligand.[1][2] It is especially common in the organometallic chemistry of transition metals with multi-atomic ligands such as carbon monoxide, ethylene or the nitrosonium cation. Electrons from the metal are used to bond to the ligand, in the process relieving the metal of excess negative charge. Compounds where π backbonding occurs include Ni(CO)4 and Zeise's salt. IUPAC offers the following definition for backbonding:
A description of the bonding of π-conjugated ligands to a transition metal which involves a synergic process with donation of electrons from the filled π-orbital or lone electron pair orbital of the ligand into an empty orbital of the metal (donor–acceptor bond), together with release (back donation) of electrons from an nd orbital of the metal (which is of π-symmetry with respect to the metal–ligand axis) into the empty π*-antibonding orbital of the ligand.[3]
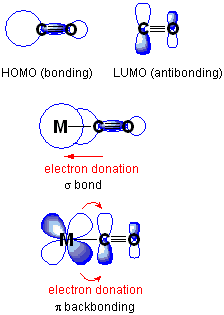
Metal carbonyls, nitrosyls, and isocyanides
The electrons are partially transferred from a d-orbital of the metal to anti-bonding molecular orbitals of CO (and its analogues). This electron-transfer (i) strengthens the metal–C bond and (ii) weakens the C–O bond. The strengthening of the M–CO bond is reflected in increases of the vibrational frequencies for the M–C bond (often outside of the range for the usual IR spectrophotometers). Furthermore, the M–CO bond length is shortened. The weakening of the C–O bond is indicated by a decrease in the wavenumber of the νCO band(s) from that for free CO (2143 cm−1), for example to 2060 cm−1 in Ni(CO)4 and 1981 cm−1 in Cr(CO)6, and 1790 cm−1 in the anion [Fe(CO)4]2−.[4] For this reason, IR spectroscopy is an important diagnostic technique in metal–carbonyl chemistry. The article infrared spectroscopy of metal carbonyls discusses this in detail.
Many ligands other than CO are strong "backbonders". Nitric oxide is an even stronger π-acceptor than is CO and νNO is a diagnostic tool in metal–nitrosyl chemistry. Isocyanides, RNC, are another class of ligands that are capable of π-backbonding. In contrast with CO, the σ-donor lone pair on the C atom of isocyanides is antibonding in nature and upon complexation the CN bond is strengthened and the νCN increased. At the same time, π-backbonding lowers the νCN. Depending on the balance of σ-bonding versus π-backbonding, the νCN can either be raised (for example, upon complexation with weak π-donor metals, such as Pt(II)) or lowered (for example, upon complexation with strong π-donor metals, such as Ni(0)). [5] For the isocyanides, an additional parameter is the MC=N–C angle, which deviates from 180° in highly electron-rich systems. Other ligands have weak π-backbonding abilities, which creates a labilization effect of CO, which is described by the cis effect.
Metal–alkene and metal–alkyne complexes
As in metal–carbonyls, electrons are partially transferred from a d-orbital of the metal to antibonding molecular orbitals of the alkenes and alkynes. This electron transfer (i) strengthens the metal–ligand bond and (ii) weakens the C–C bonds within the ligand. In the case of metal-alkenes and alkynes, the strengthening of the M–C2R4 and M–C2R2 bond is reflected in bending of the C–C–R angles which assume greater sp3 and sp2 character, respectively. Thus strong π backbonding causes a metal-alkene complex to assume the character of a metallacyclopropane. Electronegative substituents exhibit greater π backbonding. Thus, strong π backbonding ligands are tetrafluoroethylene, tetracyanoethylene, and hexafluoro-2-butyne.
Metal-phosphine complexes
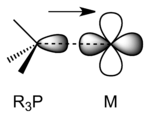
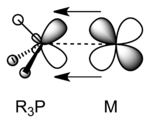
Phosphines accept electron density from metal p or d orbitals into combinations of P–C σ* antibonding orbitals that have π symmetry.[6] When phosphines bond to electron-rich metal atoms, backbonding would be expected to lengthen P–C bonds as P–C σ* orbitals become populated by electrons. The expected lengthening of the P–C distance is often hidden by an opposing effect: as the phosphorus lone pair is donated to the metal, P(lone pair)–R(bonding pair) repulsions decrease, which acts to shorten the P–C bond. The two effects have been deconvoluted by comparing the structures of pairs of metal-phosphine complexes that differ only by one electron.[7] Oxidation of R3P–M complexes results in longer M–P bonds and shorter P–C bonds, consistent with π-backbonding.[8] In early work, phosphine ligands were thought to utilize 3d orbitals to form M–P pi-bonding, but it is now accepted that d-orbitals on phosphorus are not involved in bonding as they are too high in energy.[9][10]
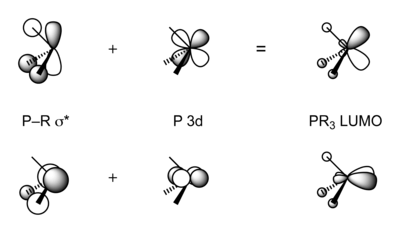
See also
- Bridging carbonyl
- Dewar–Chatt–Duncanson model
- 18-electron rule
- Ligand field theory
- Pi-donor ligands
References
- Miessler, Gary L.; Tarr, Donald Arthur (1999). Inorganic Chemistry. p. 338. ISBN 9780138418915.
- Cotton, Frank Albert; Wilkinson, Geoffrey; Murillo, Carlos A. (1999). Advanced Inorganic Chemistry. ISBN 9780471199571.
- McNaught, A. D.; Wilkinson, A. (2006). IUPAC. Compendium of Chemical Terminology, 2nd ed. (the "Gold Book"). Oxford: Blackwell Scientific Publications. doi:10.1351/goldbook. ISBN 978-0-9678550-9-7.
- Housecroft, C. E.; Sharpe, A. G. (2005). Inorganic Chemistry (2nd ed.). Pearson Prentice-Hall. p. 702. ISBN 978-0-130-39913-7.
- Crabtree, Robert H. (2014). The Organometallic Chemistry of the Transition Metals (6th ed.). Wiley. p. 105–106. ISBN 978-1-11813807-6.
- Orpen, A. G.; Connelly, N. G. (1990). "Structural systematics: the role of P–A σ* orbitals in metal–phosphorus π-bonding in redox-related pairs of M–PA3 complexes (A = R, Ar, OR; R = alkyl)". Organometallics. 9 (4): 1206–1210. doi:10.1021/om00118a048.
- Crabtree, Robert H. (2009). The Organometallic Chemistry of the Transition Metals (5th ed.). Wiley. pp. 99–100. ISBN 978-0-470-25762-3.
- Dunne, B. J.; Morris, R. B.; Orpen, A. G. (1991). "Structural systematics. Part 3. Geometry deformations in triphenylphosphine fragments: A test of bonding theories in phosphine complexes". Journal of the Chemical Society, Dalton Transactions: 653. doi:10.1039/dt9910000653.
- Gilheany, D. G. (1994). "No d Orbitals but Walsh Diagrams and Maybe Banana Bonds: Chemical Bonding in Phosphines, Phosphine Oxides, and Phosphonium Ylides". Chem. Rev. 94 (5): 1339–1374. doi:10.1021/cr00029a008.
- Fey, N.; Orpen, A. G.; Harvey, J. N. (2009). "Building ligand knowledge bases for organometallic chemistry: Computational description of phosphorus(III)-donor ligands and the metal–phosphorus bonds". Coord. Chem. Rev. 253 (5–6): 704–722. doi:10.1016/j.ccr.2008.04.017.