Alpha helix
The alpha helix (α-helix) is a common motif in the secondary structure of proteins and is a right hand-helix conformation in which every backbone N−H group hydrogen bonds to the backbone C=O group of the amino acid located three or four residues earlier along the protein sequence.
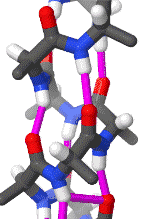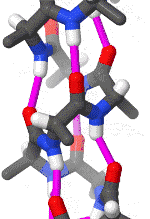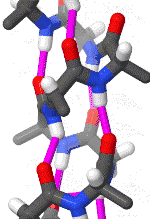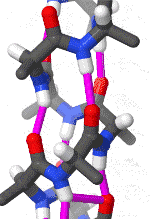
The alpha helix is also called a classic Pauling–Corey–Branson α-helix. The name 3.613-helix is also used for this type of helix, denoting the average number of residues per helical turn, with 13 atoms being involved in the ring formed by the hydrogen bond.
Among types of local structure in proteins, the α-helix is the most extreme and the most predictable from sequence, as well as the most prevalent.
.png)
Discovery
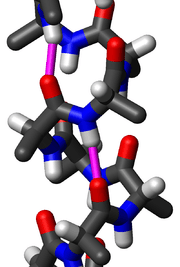
In the early 1930s, William Astbury showed that there were drastic changes in the X-ray fiber diffraction of moist wool or hair fibers upon significant stretching. The data suggested that the unstretched fibers had a coiled molecular structure with a characteristic repeat of ≈5.1 ångströms (0.51 nanometres).
Astbury initially proposed a kinked-chain structure for the fibers. He later joined other researchers (notably the American chemist Maurice Huggins) in proposing that:
- the unstretched protein molecules formed a helix (which he called the α-form)
- the stretching caused the helix to uncoil, forming an extended state (which he called the β-form).
Although incorrect in their details, Astbury's models of these forms were correct in essence and correspond to modern elements of secondary structure, the α-helix and the β-strand (Astbury's nomenclature was kept), which were developed by Linus Pauling, Robert Corey and Herman Branson in 1951 (see below); that paper showed both right- and left-handed helices, although in 1960 the crystal structure of myoglobin[1] showed that the right-handed form is the common one. Hans Neurath was the first to show that Astbury's models could not be correct in detail, because they involved clashes of atoms.[2] Neurath's paper and Astbury's data inspired H. S. Taylor,[3] Maurice Huggins[4] and Bragg and collaborators[5] to propose models of keratin that somewhat resemble the modern α-helix.
Two key developments in the modeling of the modern α-helix were: the correct bond geometry, thanks to the crystal structure determinations of amino acids and peptides and Pauling's prediction of planar peptide bonds; and his relinquishing of the assumption of an integral number of residues per turn of the helix. The pivotal moment came in the early spring of 1948, when Pauling caught a cold and went to bed. Being bored, he drew a polypeptide chain of roughly correct dimensions on a strip of paper and folded it into a helix, being careful to maintain the planar peptide bonds. After a few attempts, he produced a model with physically plausible hydrogen bonds. Pauling then worked with Corey and Branson to confirm his model before publication.[6] In 1954, Pauling was awarded his first Nobel Prize "for his research into the nature of the chemical bond and its application to the elucidation of the structure of complex substances"[7] (such as proteins), prominently including the structure of the α-helix.
Structure
Geometry and hydrogen bonding
The amino acids in an α-helix are arranged in a right-handed helical structure where each amino acid residue corresponds to a 100° turn in the helix (i.e., the helix has 3.6 residues per turn), and a translation of 1.5 Å (0.15 nm) along the helical axis. Dunitz[8] describes how Pauling's first article on the theme in fact shows a left-handed helix, the enantiomer of the true structure. Short pieces of left-handed helix sometimes occur with a large content of achiral glycine amino acids, but are unfavorable for the other normal, biological L-amino acids. The pitch of the alpha-helix (the vertical distance between consecutive turns of the helix) is 5.4 Å (0.54 nm), which is the product of 1.5 and 3.6. What is most important is that the N-H group of an amino acid forms a hydrogen bond with the C=O group of the amino acid four residues earlier; this repeated i + 4 → i hydrogen bonding is the most prominent characteristic of an α-helix. Official international nomenclature[9][10] specifies two ways of defining α-helices, rule 6.2 in terms of repeating φ, ψ torsion angles (see below) and rule 6.3 in terms of the combined pattern of pitch and hydrogen bonding. The α-helices can be identified in protein structure using several computational methods, one of which is DSSP (Define Secondary Structure of Protein).[11]
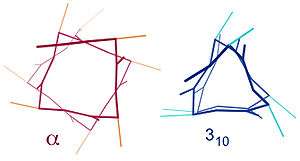
Similar structures include the 310 helix (i + 3 → i hydrogen bonding) and the π-helix (i + 5 → i hydrogen bonding). The α-helix can be described as a 3.613 helix, since the i + 4 spacing adds three more atoms to the H-bonded loop compared to the tighter 310 helix, and on average, 3.6 amino acids are involved in one ring of α-helix. The subscripts refer to the number of atoms (including the hydrogen) in the closed loop formed by the hydrogen bond.[12]
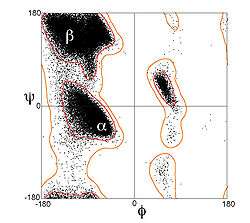
Residues in α-helices typically adopt backbone (φ, ψ) dihedral angles around (−60°, −45°), as shown in the image at right. In more general terms, they adopt dihedral angles such that the ψ dihedral angle of one residue and the φ dihedral angle of the next residue sum to roughly −105°. As a consequence, α-helical dihedral angles, in general, fall on a diagonal stripe on the Ramachandran diagram (of slope −1), ranging from (−90°, −15°) to (−35°, −70°). For comparison, the sum of the dihedral angles for a 310 helix is roughly −75°, whereas that for the π-helix is roughly −130°. The general formula for the rotation angle Ω per residue of any polypeptide helix with trans isomers is given by the equation[14][15]
- 3 cos Ω = 1 − 4 cos2 φ + ψ/2
The α-helix is tightly packed; there is almost no free space within the helix. The amino-acid side-chains are on the outside of the helix, and point roughly "downward" (i.e., toward the N-terminus), like the branches of an evergreen tree (Christmas tree effect). This directionality is sometimes used in preliminary, low-resolution electron-density maps to determine the direction of the protein backbone.[16]
Stability
Helices observed in proteins can range from four to over forty residues long, but a typical helix contains about ten amino acids (about three turns). In general, short polypeptides do not exhibit much α-helical structure in solution, since the entropic cost associated with the folding of the polypeptide chain is not compensated for by a sufficient amount of stabilizing interactions. In general, the backbone hydrogen bonds of α-helices are considered slightly weaker than those found in β-sheets, and are readily attacked by the ambient water molecules. However, in more hydrophobic environments such as the plasma membrane, or in the presence of co-solvents such as trifluoroethanol (TFE), or isolated from solvent in the gas phase,[17] oligopeptides readily adopt stable α-helical structure. Furthermore, crosslinks can be incorporated into peptides to conformationally stabilize helical folds. Crosslinks stabilize the helical state by entropically destabilizing the unfolded state and by removing enthalpically stabilized "decoy" folds that compete with the fully helical state.[18] It has been shown that α-helices are more stable, robust to mutations and designable than β-strands in natural proteins,[19] and also in artificial designed proteins.[20]
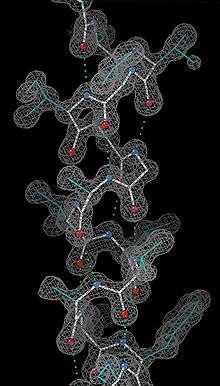
Experimental determination
Since the α-helix is defined by its hydrogen bonds and backbone conformation, the most detailed experimental evidence for α-helical structure comes from atomic-resolution X-ray crystallography such as the example shown at right. It is clear that all the backbone carbonyl oxygens point downward (toward the C-terminus) but splay out slightly, and the H-bonds are approximately parallel to the helix axis. Protein structures from NMR spectroscopy also show helices well, with characteristic observations of nuclear Overhauser effect (NOE) couplings between atoms on adjacent helical turns. In some cases, the individual hydrogen bonds can be observed directly as a small scalar coupling in NMR.
There are several lower-resolution methods for assigning general helical structure. The NMR chemical shifts (in particular of the Cα, Cβ and C′) and residual dipolar couplings are often characteristic of helices. The far-UV (170–250 nm) circular dichroism spectrum of helices is also idiosyncratic, exhibiting a pronounced double minimum at around 208 and 222 nm. Infrared spectroscopy is rarely used, since the α-helical spectrum resembles that of a random coil (although these might be discerned by, e.g., hydrogen-deuterium exchange). Finally, cryo electron microscopy is now capable of discerning individual α-helices within a protein, although their assignment to residues is still an active area of research.
Long homopolymers of amino acids often form helices if soluble. Such long, isolated helices can also be detected by other methods, such as dielectric relaxation, flow birefringence, and measurements of the diffusion constant. In stricter terms, these methods detect only the characteristic prolate (long cigar-like) hydrodynamic shape of a helix, or its large dipole moment.
Amino-acid propensities
Different amino-acid sequences have different propensities for forming α-helical structure. Methionine, alanine, leucine, glutamate, and lysine uncharged ("MALEK" in the amino-acid 1-letter codes) all have especially high helix-forming propensities, whereas proline and glycine have poor helix-forming propensities.[21] Proline either breaks or kinks a helix, both because it cannot donate an amide hydrogen bond (having no amide hydrogen), and also because its sidechain interferes sterically with the backbone of the preceding turn – inside a helix, this forces a bend of about 30° in the helix's axis.[12] However, proline is often seen as the first residue of a helix, it is presumed due to its structural rigidity. At the other extreme, glycine also tends to disrupt helices because its high conformational flexibility makes it entropically expensive to adopt the relatively constrained α-helical structure.
Table of standard amino acid alpha-helical propensities
Estimated differences in free energy, Δ(ΔG), estimated in kcal/mol per residue in an α-helical configuration, relative to alanine arbitrarily set as zero. Higher numbers (more positive free energies) are less favoured. Significant deviations from these average numbers are possible, depending on the identities of the neighbouring residues.
Differences in free energy per residue[22] Amino acid 3-
letter1-
letterHelical penalty kcal/mol kJ/mol Alanine Ala A 0.00 0.00 Arginine Arg R 0.21 0.88 Asparagine Asn N 0.65 2.72 Aspartic acid Asp D 0.69 2.89 Cysteine Cys C 0.68 2.85 Glutamic acid Glu E 0.40 1.67 Glutamine Gln Q 0.39 1.63 Glycine Gly G 1.00 4.18 Histidine His H 0.61 2.55 Isoleucine Ile I 0.41 1.72 Leucine Leu L 0.21 0.88 Lysine Lys K 0.26 1.09 Methionine Met M 0.24 1.00 Phenylalanine Phe F 0.54 2.26 Proline Pro P 3.16 13.22 Serine Ser S 0.50 2.09 Threonine Thr T 0.66 2.76 Tryptophan Trp W 0.49 2.05 Tyrosine Tyr Y 0.53 2.22 Valine Val V 0.61 2.55
Dipole moment
A helix has an overall dipole moment due to the aggregate effect of the individual microdipoles from the carbonyl groups of the peptide bond pointing along the helix axis.[23] The effects of this macrodipole are a matter of some controversy. α-helices often occur with the N-terminal end bound by a negatively charged group, sometimes an amino acid side chain such as glutamate or aspartate, or sometimes a phosphate ion. Some regard the helix macrodipole as interacting electrostatically with such groups. Others feel that this is misleading and it is more realistic to say that the hydrogen bond potential of the free NH groups at the N-terminus of an α-helix can be satisfied by hydrogen bonding; this can also be regarded as set of interactions between local microdipoles such as C=O···H−N.[24][25]
Coiled coils
Coiled-coil α helices are highly stable forms in which two or more helices wrap around each other in a "supercoil" structure. Coiled coils contain a highly characteristic sequence motif known as a heptad repeat, in which the motif repeats itself every seven residues along the sequence (amino acid residues, not DNA base-pairs). The first and especially the fourth residues (known as the a and d positions) are almost always hydrophobic; the fourth residue is typically leucine – this gives rise to the name of the structural motif called a leucine zipper, which is a type of coiled-coil. These hydrophobic residues pack together in the interior of the helix bundle. In general, the fifth and seventh residues (the e and g positions) have opposing charges and form a salt bridge stabilized by electrostatic interactions. Fibrous proteins such as keratin or the "stalks" of myosin or kinesin often adopt coiled-coil structures, as do several dimerizing proteins. A pair of coiled-coils – a four-helix bundle – is a very common structural motif in proteins. For example, it occurs in human growth hormone and several varieties of cytochrome. The Rop protein, which promotes plasmid replication in bacteria, is an interesting case in which a single polypeptide forms a coiled-coil and two monomers assemble to form a four-helix bundle.
Facial arrangements
The amino acids that make up a particular helix can be plotted on a helical wheel, a representation that illustrates the orientations of the constituent amino acids (see the article for leucine zipper for such a diagram). Often in globular proteins, as well as in specialized structures such as coiled-coils and leucine zippers, an α-helix will exhibit two "faces" – one containing predominantly hydrophobic amino acids oriented toward the interior of the protein, in the hydrophobic core, and one containing predominantly polar amino acids oriented toward the solvent-exposed surface of the protein.
Changes in binding orientation also occur for facially-organized oligopeptides. This pattern is especially common in antimicrobial peptides, and many models have been devised to describe how this relates to their function. Common to many of them is that the hydrophobic face of the antimicrobial peptide forms pores in the plasma membrane after associating with the fatty chains at the membrane core.[26][27]
Larger-scale assemblies
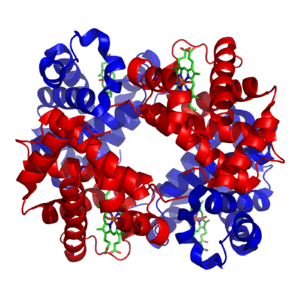
Myoglobin and hemoglobin, the first two proteins whose structures were solved by X-ray crystallography, have very similar folds made up of about 70% α-helix, with the rest being non-repetitive regions, or "loops" that connect the helices. In classifying proteins by their dominant fold, the Structural Classification of Proteins database maintains a large category specifically for all-α proteins.
Hemoglobin then has an even larger-scale quaternary structure, in which the functional oxygen-binding molecule is made up of four subunits.
Functional roles
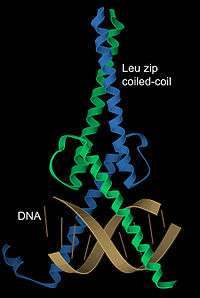
DNA binding
α-Helices have particular significance in DNA binding motifs, including helix-turn-helix motifs, leucine zipper motifs and zinc finger motifs. This is because of the convenient structural fact that the diameter of an α-helix is about 12 Å (1.2 nm) including an average set of sidechains, about the same as the width of the major groove in B-form DNA, and also because coiled-coil (or leucine zipper) dimers of helices can readily position a pair of interaction surfaces to contact the sort of symmetrical repeat common in double-helical DNA.[28] An example of both aspects is the transcription factor Max (see image at left), which uses a helical coiled coil to dimerize, positioning another pair of helices for interaction in two successive turns of the DNA major groove.
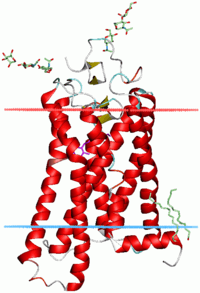
Membrane spanning
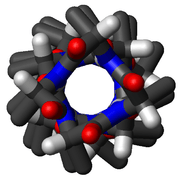
α-Helices are also the most common protein structure element that crosses biological membranes (transmembrane protein),[29] it is presumed because the helical structure can satisfy all backbone hydrogen-bonds internally, leaving no polar groups exposed to the membrane if the sidechains are hydrophobic. Proteins are sometimes anchored by a single membrane-spanning helix, sometimes by a pair, and sometimes by a helix bundle, most classically consisting of seven helices arranged up-and-down in a ring such as for rhodopsins (see image at right) or for G protein–coupled receptors (GPCRs).
Mechanical properties
α-Helices under axial tensile deformation, a characteristic loading condition that appears in many alpha-helix-rich filaments and tissues, results in a characteristic three-phase behavior of stiff-soft-stiff tangent modulus.[30] Phase I corresponds to the small-deformation regime during which the helix is stretched homogeneously, followed by phase II, in which alpha-helical turns break mediated by the rupture of groups of H-bonds. Phase III is typically associated with large-deformation covalent bond stretching.
Dynamical features
Alpha-helices in proteins may have low-frequency accordion-like motion as observed by the Raman spectroscopy[31] and analyzed via the quasi-continuum model.[32][33] Helices not stabilized by tertiary interactions show dynamic behavior, which can be mainly attributed to helix fraying from the ends.[34]
Helix–coil transition
Homopolymers of amino acids (such as polylysine) can adopt α-helical structure at low temperature that is "melted out" at high temperatures. This helix–coil transition was once thought to be analogous to protein denaturation. The statistical mechanics of this transition can be modeled using an elegant transfer matrix method, characterized by two parameters: the propensity to initiate a helix and the propensity to extend a helix.
In art

At least five artists have made explicit reference to the α-helix in their work: Julie Newdoll in painting and Julian Voss-Andreae, Bathsheba Grossman, Byron Rubin, and Mike Tyka in sculpture.
San Francisco area artist Julie Newdoll,[35] who holds a degree in Microbiology with a minor in art, has specialized in paintings inspired by microscopic images and molecules since 1990. Her painting "Rise of the Alpha Helix" (2003) features human figures arranged in an α helical arrangement. According to the artist, "the flowers reflect the various types of sidechains that each amino acid holds out to the world".[35] This same metaphor is also echoed from the scientist's side: "β sheets do not show a stiff repetitious regularity but flow in graceful, twisting curves, and even the α-helix is regular more in the manner of a flower stem, whose branching nodes show the influence of environment, developmental history, and the evolution of each part to match its own idiosyncratic function."[12]
Julian Voss-Andreae is a German-born sculptor with degrees in experimental physics and sculpture. Since 2001 Voss-Andreae creates "protein sculptures"[36] based on protein structure with the α-helix being one of his preferred objects. Voss-Andreae has made α-helix sculptures from diverse materials including bamboo and whole trees. A monument Voss-Andreae created in 2004 to celebrate the memory of Linus Pauling, the discoverer of the α-helix, is fashioned from a large steel beam rearranged in the structure of the α-helix. The 10-foot-tall (3 m), bright-red sculpture stands in front of Pauling's childhood home in Portland, Oregon.
Ribbon diagrams of α-helices are a prominent element in the laser-etched crystal sculptures of protein structures created by artist Bathsheba Grossman, such as those of insulin, hemoglobin, and DNA polymerase.[37] Byron Rubin is a former protein crystallographer now professional sculptor in metal of proteins, nucleic acids, and drug molecules – many of which featuring α-helices, such as subtilisin, human growth hormone, and phospholipase A2.[38]
Mike Tyka is a computational biochemist at the University of Washington working with David Baker. Tyka has been making sculptures of protein molecules since 2010 from copper and steel, including ubiquitin and a potassium channel tetramer.[39]
See also
- 310 helix
- Pi helix
- Beta sheet
- Davydov soliton
- Folding (chemistry)
- Knobs into holes packing
- Proteopedia Helices_in_Proteins
References
- Kendrew JC, Dickerson RE, Strandberg BE, Hart RG, Davies DR, Phillips DC, Shore VC (February 1960). "Structure of myoglobin: A three-dimensional Fourier synthesis at 2 Å resolution". Nature. 185 (4711): 422–7. Bibcode:1960Natur.185..422K. doi:10.1038/185422a0. PMID 18990802. S2CID 4167651.
- Neurath H (1940). "Intramolecular folding of polypeptide chains in relation to protein structure". Journal of Physical Chemistry. 44 (3): 296–305. doi:10.1021/j150399a003.
- Taylor HS (1942). "Large molecules through atomic spectacles". Proceedings of the American Philosophical Society. 85 (1): 1–12. JSTOR 985121.
- Huggins M (1943). "The structure of fibrous proteins". Chemical Reviews. 32 (2): 195–218. doi:10.1021/cr60102a002.
- Bragg WL, Kendrew JC, Perutz MF (1950). "Polypeptide chain configurations in crystalline proteins". Proceedings of the Royal Society of London. Series A. Mathematical and Physical Sciences. 203 (1074): 321–?. Bibcode:1950RSPSA.203..321B. doi:10.1098/rspa.1950.0142. S2CID 93804323.
- Pauling L, Corey RB, Branson HR (April 1951). "The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain". Proceedings of the National Academy of Sciences of the United States of America. 37 (4): 205–11. Bibcode:1951PNAS...37..205P. doi:10.1073/pnas.37.4.205. PMC 1063337. PMID 14816373.
- "The Nobel Prize in Chemistry 1954".
- Dunitz J (2001). "Pauling's Left-Handed α-Helix". Angewandte Chemie International Edition. 40 (22): 4167–4173. doi:10.1002/1521-3773(20011119)40:22<4167::AID-ANIE4167>3.0.CO;2-Q. PMID 29712120.
- IUPAC-IUB Commission on Biochemical Nomenclature (1970). "Abbreviations and symbols for the description of the conformation of polypeptide chains". Journal of Biological Chemistry. 245: 6489–6497.
- "Polypeptide Conformations 1 and 2". www.sbcs.qmul.ac.uk. Retrieved 5 November 2018.
- Kabsch W, Sander C (December 1983). "Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features". Biopolymers. 22 (12): 2577–637. doi:10.1002/bip.360221211. PMID 6667333.
- Richardson JS (1981). "The anatomy and taxonomy of protein structure". Advances in Protein Chemistry. 34: 167–339. doi:10.1016/S0065-3233(08)60520-3. ISBN 9780120342341. PMID 7020376.
- Lovell SC, Davis IW, Arendall WB, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC (February 2003). "Structure validation by Calpha geometry: phi,psi and Cbeta deviation". Proteins. 50 (3): 437–50. doi:10.1002/prot.10286. PMID 12557186.
- Dickerson RE, Geis I (1969), Structure and Action of Proteins, Harper, New York
- Zorko, Matjaž (2010). "Structural Organization of Proteins". In Langel, Ülo; Cravatt, Benjamin F.; Gräslund, Astrid; von Heijne, Gunnar; Land, Tiit; Niessen, Sherry; Zorko, Matjaž (eds.). Introduction to Peptides and Proteins. Boca Raton: CRC Press. pp. 36–57. ISBN 9781439882047.
- Terwilliger TC (March 2010). "Rapid model building of alpha-helices in electron-density maps". Acta Crystallographica Section D. 66 (Pt 3): 268–75. doi:10.1107/S0907444910000314. PMC 2827347. PMID 20179338.
- Hudgins RR, Jarrold MF (1999). "Helix Formation in Unsolvated Alanine-Based Peptides: Helical Monomers and Helical Dimers". Journal of the American Chemical Society. 121 (14): 3494–3501. doi:10.1021/ja983996a.
- Kutchukian PS, Yang JS, Verdine GL, Shakhnovich EI (April 2009). "All-atom model for stabilization of alpha-helical structure in peptides by hydrocarbon staples". Journal of the American Chemical Society. 131 (13): 4622–7. doi:10.1021/ja805037p. PMC 2735086. PMID 19334772.
- Abrusan G, Marsh JA (2016). "Alpha helices are more robust to mutations than beta strands". PLOS Computational Biology. 12 (12): e1005242. Bibcode:2016PLSCB..12E5242A. doi:10.1371/journal.pcbi.1005242. PMC 5147804. PMID 27935949.
- Rocklin GJ, et al. (2017). "Global analysis of protein folding using massively parallel design, synthesis, and testing". Science. 357 (6347): 168–175. Bibcode:2017Sci...357..168R. doi:10.1126/science.aan0693. PMC 5568797. PMID 28706065.
- Pace CN, Scholtz JM (July 1998). "A helix propensity scale based on experimental studies of peptides and proteins". Biophysical Journal. 75 (1): 422–7. Bibcode:1998BpJ....75..422N. doi:10.1016/S0006-3495(98)77529-0. PMC 1299714. PMID 9649402.
- Pace, C. Nick; Scholtz, J. Martin (1998). "A Helix Propensity Scale Based on Experimental Studies of Peptides and Proteins". Biophysical Journal. 75. pp. 422–427. Bibcode:1998BpJ....75..422N. doi:10.1016/s0006-3495(98)77529-0.
- Hol WG, van Duijnen PT, Berendsen HJ (1978). "The alpha helix dipole and the properties of proteins". Nature. 273 (5662): 443–446. Bibcode:1978Natur.273..443H. doi:10.1038/273443a0. PMID 661956. S2CID 4147335.
- He JJ, Quiocho FA (October 1993). "Dominant role of local dipoles in stabilizing uncompensated charges on a sulfate sequestered in a periplasmic active transport protein". Protein Science. 2 (10): 1643–7. doi:10.1002/pro.5560021010. PMC 2142251. PMID 8251939.
- Milner-White EJ (November 1997). "The partial charge of the nitrogen atom in peptide bonds". Protein Science. 6 (11): 2477–82. doi:10.1002/pro.5560061125. PMC 2143592. PMID 9385654.
- Kohn, Eric M.; Shirley, David J.; Arotsky, Lubov; Picciano, Angela M.; Ridgway, Zachary; Urban, Michael W.; Carone, Benjamin R.; Caputo, Gregory A. (2018-02-04). "Role of Cationic Side Chains in the Antimicrobial Activity of C18G". Molecules. 23 (2): 329. doi:10.3390/molecules23020329. PMC 6017431. PMID 29401708.
- Toke, Orsolya (2005). "Antimicrobial peptides: new candidates in the fight against bacterial infections". Biopolymers. 80 (6): 717–735. doi:10.1002/bip.20286. ISSN 0006-3525. PMID 15880793.
- Branden & Tooze, chapter 10
- Branden & Tooze, chapter 12.
- Ackbarow T, Chen X, Keten S, Buehler MJ (October 2007). "Hierarchies, multiple energy barriers, and robustness govern the fracture mechanics of alpha-helical and beta-sheet protein domains". Proceedings of the National Academy of Sciences of the United States of America. 104 (42): 16410–5. Bibcode:2007PNAS..10416410A. doi:10.1073/pnas.0705759104. PMC 2034213. PMID 17925444.
- Painter PC, Mosher LE, Rhoads C (July 1982). "Low-frequency modes in the Raman spectra of proteins". Biopolymers. 21 (7): 1469–72. doi:10.1002/bip.360210715. PMID 7115900.
- Chou KC (December 1983). "Identification of low-frequency modes in protein molecules". The Biochemical Journal. 215 (3): 465–9. doi:10.1042/bj2150465. PMC 1152424. PMID 6362659.
- Chou KC (May 1984). "Biological functions of low-frequency vibrations (phonons). III. Helical structures and microenvironment". Biophysical Journal. 45 (5): 881–9. Bibcode:1984BpJ....45..881C. doi:10.1016/S0006-3495(84)84234-4. PMC 1434967. PMID 6428481.
- Fierz B, Reiner A, Kiefhaber T (January 2009). "Local conformational dynamics in alpha-helices measured by fast triplet transfer". Proceedings of the National Academy of Sciences of the United States of America. 106 (4): 1057–62. Bibcode:2009PNAS..106.1057F. doi:10.1073/pnas.0808581106. PMC 2633579. PMID 19131517.
- "Julie Newdoll Scientifically Inspired Art, Music, Board Games". www.brushwithscience.com. Retrieved 2016-04-06.
- Voss-Andreae J (2005). "Protein Sculptures: Life's Building Blocks Inspire Art". Leonardo. 38: 41–45. doi:10.1162/leon.2005.38.1.41. S2CID 57558522.
- Grossman, Bathsheba. "About the Artist". Bathsheba Sculpture. Retrieved 2016-04-06.
- "About". molecularsculpture.com. Retrieved 2016-04-06.
- Tyka, Mike. "About". www.miketyka.com. Retrieved 2016-04-06.
Further reading
- Tooze J, Brändén C (1999). Introduction to protein structure. New York: Garland Pub. ISBN 0-8153-2304-2..
- Eisenberg D (September 2003). "The discovery of the alpha-helix and beta-sheet, the principal structural features of proteins". Proceedings of the National Academy of Sciences of the United States of America. 100 (20): 11207–10. Bibcode:2003PNAS..10011207E. doi:10.1073/pnas.2034522100. PMC 208735. PMID 12966187.
- Astbury WT, Woods HJ (1931). "The Molecular Weights of Proteins". Nature. 127 (3209): 663–665. Bibcode:1931Natur.127..663A. doi:10.1038/127663b0. S2CID 4133226.
- Astbury WT, Street A (1931). "X-ray studies of the structures of hair, wool and related fibres. I. General". Trans. R. Soc. Lond. A230: 75–101. Bibcode:1932RSPTA.230...75A. doi:10.1098/rsta.1932.0003.
- Astbury WT (1933). "Some Problems in the X-ray Analysis of the Structure of Animal Hairs and Other Protein Fibers". Trans. Faraday Soc. 29 (140): 193–211. doi:10.1039/tf9332900193.
- Astbury WT, Woods HJ (1934). "X-ray studies of the structures of hair, wool and related fibres. II. The molecular structure and elastic properties of hair keratin". Philosophical Transactions of the Royal Society of London Series A. 232 (707–720): 333–394. Bibcode:1934RSPTA.232..333A. doi:10.1098/rsta.1934.0010.
- Astbury WT, Sisson WA (1935). "X-ray studies of the structures of hair, wool and related fibres. III. The configuration of the keratin molecule and its orientation in the biological cell". Proceedings of the Royal Society. A150 (871): 533–551. Bibcode:1935RSPSA.150..533A. doi:10.1098/rspa.1935.0121.
- Sugeta H, Miyazawa T (1967). "General Method for Calculating Helical Parameters of Polymer Chains from Bond Lengths, Bond Angles, and Internal-Rotation Angles". Biopolymers. 5 (7): 673–679. doi:10.1002/bip.1967.360050708.
- Wada A (1976). "The alpha-helix as an electric macro-dipole". Advances in Biophysics: 1–63. PMID 797240.
- Chothia C, Levitt M, Richardson D (October 1977). "Structure of proteins: packing of alpha-helices and pleated sheets". Proceedings of the National Academy of Sciences of the United States of America. 74 (10): 4130–4. Bibcode:1977PNAS...74.4130C. doi:10.1073/pnas.74.10.4130. PMC 431889. PMID 270659.
- Chothia C, Levitt M, Richardson D (January 1981). "Helix to helix packing in proteins". Journal of Molecular Biology. 145 (1): 215–50. doi:10.1016/0022-2836(81)90341-7. PMID 7265198.
- Hol WG (1985). "The role of the alpha-helix dipole in protein function and structure". Progress in Biophysics and Molecular Biology. 45 (3): 149–95. doi:10.1016/0079-6107(85)90001-X. PMID 3892583.
- Barlow DJ, Thornton JM (June 1988). "Helix geometry in proteins". Journal of Molecular Biology. 201 (3): 601–19. doi:10.1016/0022-2836(88)90641-9. PMID 3418712.
- Murzin AG, Finkelstein AV (December 1988). "General architecture of the alpha-helical globule". Journal of Molecular Biology. 204 (3): 749–69. doi:10.1016/0022-2836(88)90366-X. PMID 3225849.
External links
- NetSurfP ver. 1.1 - Protein Surface Accessibility and Secondary Structure Predictions
- α-helix rotational angle calculator
- Artist Julie Newdoll's website
- Artist Julian Voss-Andreae's website
| Curves |  | ||
|---|---|---|---|
| Helices |
| ||
| Spirals | |||