Wolff rearrangement
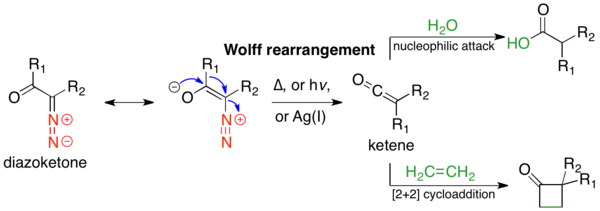
The Wolff rearrangement is a reaction in organic chemistry in which an α-diazocarbonyl compound is converted into a ketene by loss of dinitrogen with accompanying 1,2-rearrangement. The Wolff rearrangement yields a ketene as an intermediate product, which can undergo nucleophilic attack with weakly acidic nucleophiles such as water, alcohols, and amines, to generate carboxylic acid derivatives or undergo [2+2] cycloaddition reactions to form four-membered rings.[1] The mechanism of the Wolff rearrangement has been the subject of debate since its first use. No single mechanism sufficiently describes the reaction, and there are often competing concerted and carbene-mediated pathways; for simplicity, only the textbook, concerted mechanism is shown below.[2] The reaction was discovered by Ludwig Wolff in 1902.[3] The Wolff rearrangement has great synthetic utility due to the accessibility of α-diazocarbonyl compounds, variety of reactions from the ketene intermediate, and stereochemical retention of the migrating group.[2] However, the Wolff rearrangement has limitations due to the highly reactive nature of α-diazocarbonyl compounds, which can undergo a variety of competing reactions.[1]
| Wolff rearrangement | |
|---|---|
| Named after | Ludwig Wolff |
| Reaction type | Rearrangement reaction |
| Identifiers | |
| Organic Chemistry Portal | wolff-rearrangement |
| RSC ontology ID | RXNO:0000051 |
The Wolff rearrangement can be induced via thermolysis,[3] photolysis,[4] or transition metal catalysis.[3] In this last case, the reaction is sensitive to the transition metal; silver (I) oxide or other Ag(I) catalysts work well and are generally used. The Wolff rearrangement has been used in many total syntheses; the most common use is trapping the ketene intermediate with nucleophiles to form carboxylic acid derivatives. The Arndt-Eistert homologation is a specific example of this use, wherein a carboxylic acid may be elongated by a methylene unit. Another common use is in ring-contraction methods; if the α-diazo ketone is cyclic, the Wolff rearrangement results in a ring-contracted product. The Wolff rearrangement works well in generating ring-strained systems, where other reactions may fail.
History
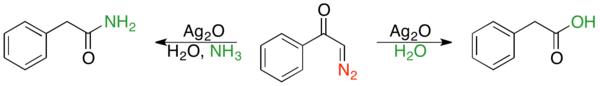
In 1902, Wolff discovered that treating diazoacetophenone with silver (I) oxide and water resulted in formation of phenylacetic acid. Similarly, treatment with silver (I) oxide and ammonia formed phenylacetamide.[3] A few years later, in an independent study, Schröter observed similar results.[5] The reaction is occasionally called the Wolff-Schröter rearrangement.[2] The Wolff rearrangement was not commonly used until 20 years after it was discovered, as facile diazo ketone synthesis was unknown until the 1930s.[2] The reaction has proven useful in synthetic organic chemistry and many reviews have been published.[1][2]
Mechanism
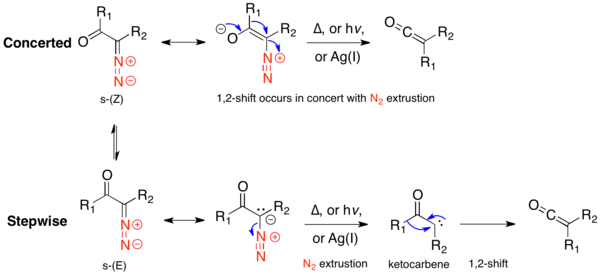
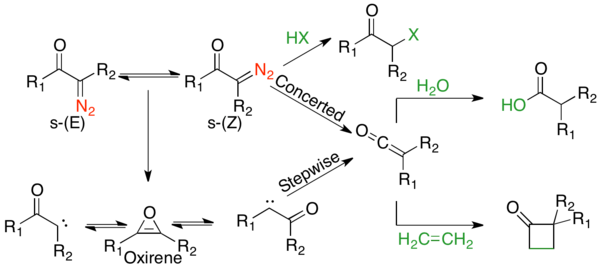
The mechanistic pathway of the Wolff-rearrangement has been the subject of much debate, as there are often competing concerted and stepwise mechanisms.[2] However, two aspects of the mechanism can be agreed upon. First, α-diazocarbonyl compounds are in an equilibrium of s-cis and s-trans-conformers, the distribution of which may influence the mechanism of the reaction. Generally, under photolysis, compounds in the s-cis conformation react in a concerted manner due to the antiperiplanar relationship between the leaving and migrating groups, whereas compounds in the s-trans conformation react stepwise through a carbene intermediate or do not rearrange. Second, regardless of the reaction mechanism, the rearrangement gives a ketene intermediate, which can be trapped by a weakly acidic nucleophile, such as an alcohol or amine, to give the corresponding ester or amide, or an olefin, to give a [2+2] cycloaddition adduct. Strong acids do not rearrange, but rather protonate the α-carbon and give SN2 products.
Stereochemistry of α-diazo ketones
Understanding the stereochemistry of α-diazo ketones is essential in elucidating the mechanism of the Wolff rearrangement. α-diazocarbonyl compounds are generally locally planar, with large rotational barriers (55–65 kJ/mol) due to C-C olefin character between the carbonyl and α-carbon, illustrated in the rightmost resonance structure.[6] Such a large barrier slows molecular rotations sufficiently to lead to an equilibrium between two conformers, an s-trans and s-cis-conformer. s-cis-Conformers are electronically favored due to Coulombic attraction between the oxygen with a partial negative charge and the cationic nitrogen, as seen in the rightmost resonance structure.[1] If R1 is large and R2 is hydrogen, s-cis is sterically favored. If R1 and R2 are large, s-trans is sterically favored; if both substituents are sufficiently large, the steric repulsion can outweigh the Coulombic attraction, leading to a preference for s-trans. Small and medium cyclic substrates are constrained in the s-cis conformation.
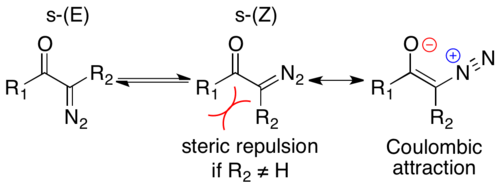
Concerted mechanism
When the α-diazo ketone is in the s-cis conformation, the leaving group (N2) and the migrating group (R1) are antiperiplanar, which favors a concerted mechanism, in which nitrogen extrusion occurs concurrently with 1,2-alkyl shift. There is evidence this mechanism occurs in both thermolytic and photolytic methods, when the s-cis-conformer is strongly favored.[7]
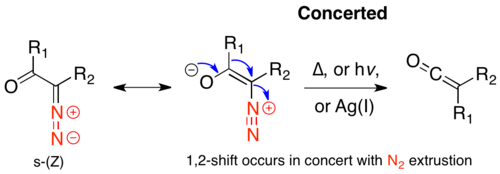
CIDNP studies show that photochemical rearrangement of diazoacetone, which largely exists in the s-cis-conformer, is concerted.[8] Product ratios from direct and triplet-sensitized photolysis have been used as evidence for proposals that claim that concerted products arise from the s-cis-conformer and stepwise products occur through the s-trans-conformer.[9]
Stepwise mechanism
s-trans-α-Diazo ketones do not have an antiperiplanar relationship between the leaving and migrating group, and thus are thought to generally rearrange stepwise. The stepwise mechanism begins with nitrogen extrusion, forming an α-ketocarbene. The α-ketocarbene can either undergo a 1,2-alkyl shift, to give the ketene product, or can undergo a 4π electrocyclic ring closure, to form an antiaromatic oxirene. This oxirene can reopen in two ways, to either α-ketocarbene, which can then form the ketene product.
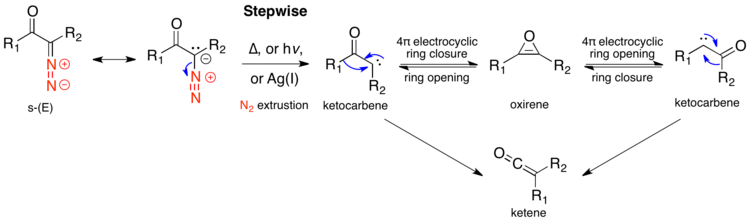
There are two primary arguments for stepwise mechanisms. The first is that rate constants of Wolff rearrangements depend on the stability of the formed carbene, rather than the migratory aptitude of the migrating group.[10] The most definitive evidence is isotopic scrambling of the ketene, as predicted by an oxirene intermediate, which can only occur in the stepwise path. In the scheme below, the red carbon is 13C labelled. The symmetric oxirene intermediate can open either way, scrambling the 13C label. If the substituents R1 and R2 are the same, one can quantify the ratio of products stemming from the concerted and stepwise mechanisms; if the substituents are different, the oxirene will have a preference in the direction it opens, and a ratio cannot be quantified, but any scrambling indicates some reactant is going through a stepwise mechanism.[1] In photolysis of diazo acetaldehyde, 8% of the label is scrambled, indicating that 16% of product is formed via the oxirene intermediate.[11] Under photolysis, the biphenyl (R1=R2=phenyl) substrate shows 20–30% label migration, implying 40–60% of product goes through the oxirene intermediate.[12] α-diazocyclohexanone shows no label scrambling under photolytic conditions, as it is entirely s-cis, and thus all substrate goes through the concerted mechanism, avoiding the oxirene intermediate.[13]
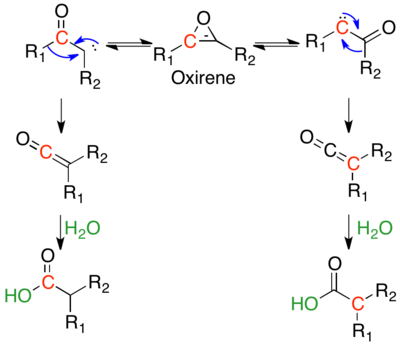
Isotopic labeling studies have been used extensively to measure the ratio of product stemming from a concerted mechanism versus a stepwise mechanism.[14] These studies confirm that reactants that prefer s-trans conformations tend to undergo stepwise reaction. The degree of scrambling is also affected by carbene stability, migratory abilities, and nucleophilicity of solvent. The observation that the migratory ability of a substituent is inversely proportional to amount of carbene formed, indicates that under photolysis, there are competing pathways for many Wolff reactions.[14] The only Wolff rearrangements that show no scrambling are s-cis constrained cyclic α-diazo ketones.[13]
Mechanistic conclusion
Under both thermolytic and photolytic conditions, there exist competing concerted and stepwise mechanisms. Many mechanistic studies have been carried out, including conformational, sensitization, kinetic, and isotopic scrambling studies. These all point to competing mechanisms, with general trends. α-Diazo ketones that exist in the s-cis conformation generally undergo a concerted mechanism, whereas those in the s-trans conformation undergo a stepwise mechanism.[1] α-diazo ketones with better migratory groups prefer a concerted mechanism.[1] However, for all substrates except cyclic α-diazo ketones that exist solely in the s-cis conformation, products come from a combination of both pathways.[1] Transition metal mediated reactions are quite varied; however, they generally prefer forming the metal carbene intermediate.[2] The complete mechanism under photolysis can be approximated in the following figure:
Migratory trends
The mechanism of the Wolff rearrangement is dependent on the aptitude of the migratory group. Migratory abilities have been determined by competition studies. In general, hydrogen migrates the fastest, and alkyl and aryl groups migrate at approximately the same rate, with alkyl migrations favored under photolysis, and aryl migrations preferred under thermolysis.[15] Substituent effects on aryl groups are negligible, with the exception of NO2, which is a poor migrator.[15] In competition studies, electron deficient alkyl, aryl, and carbonyl groups cannot compete with other migrating groups, but are still competent.[16][17][18] Heteroatoms, in general, are poor migratory groups, because their ability to donate electron density from their p orbitals into the π* C=O bond decreases migratory ability.[1] The trend is as follows:[1]
Photochemical reactions: H > alkyl ≥ aryl >> SR > OR ≥ NR2
Thermal reactions H > aryl ≥ alkyl (heteroatoms do not migrate)
Preparation of α-diazocarbonyl compounds
While known since 1902, the Wolff rearrangement did not become synthetically useful until the early 1930s, when efficient methods became available to synthesize α-diazocarbonyl compounds. The primary ways to prepare these substrates today are via the Arndt-Eistert procedure, the Franzen modification to the Dakin-West reaction, and diazo-transfer methods.
Arndt-Eistert procedure
The Arndt–Eistert reaction[19] involves the acylation of diazomethane with an acid chloride, to yield a primary α-diazo ketone. The carbon terminus of diazomethane adds to the carbonyl, to create a tetrahedral intermediate, which eliminates chloride. The chloride then deprotonates the intermediate to give the α-diazo ketone product.
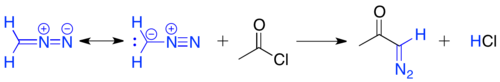
These α-diazo ketones are unstable under acidic conditions, as the α-carbon can be protonated by HCl and SN2 displacement of nitrogen can occur by chloride.
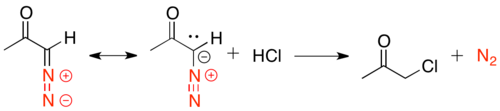
Franzen modification to Dakin-West reaction
The Dakin–West reaction is a reaction of an amino acid with an acid anhydride in the presence of a base to form keto-amides. The Franzen modification[20] to the Dakin–West reaction[21] is a more effective way to make secondary α-diazo ketones. The Franzen modification nitrosates the keto-amide with N2O3 in acetic acid, and the resulting product reacts with methoxide in methanol to give the secondary α-diazo ketone.

Diazo-transfer reactions
Diazo-transfer reactions are commonly used methods, in which an organic azide, usually tosylazide, and an activated methylene (i.e. a methylene with two withdrawing groups) react in the presence of a base to give an α-diazo-1,3-diketone.[22] The base deprotonates the methylene, yielding an enolate, which reacts with tosylazide and subsequently decomposes in the presence of a weak acid, to give the α-diazo-1,3-diketone.

The necessary requirement of two electron withdrawing groups makes this reaction one of limited scope. The scope can be broadened to substrates containing one electron withdrawing group by formylating a ketone via a Claisen condensation, followed by diazo-transfer and deformylative group transfer.[23]
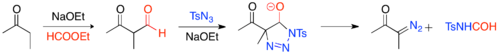
One of the greatest advantages of this method is its compatibility with unsaturated ketones. However, to achieve kinetic regioselectivity in enolate formation and greater compatibility with unsaturated carbonyls, one can induce enolate formation with lithium hexamethyldisilazide and subsequently trifluoroacylate rather than formylate.[24]
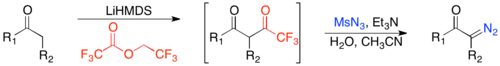
Methods for inducing rearrangement
Wolff rearrangements can be induced under thermolytic,[3] photolytic,[4] and transition-metal-catalyzed conditions.[3]
Thermal conditions to induce rearrangement require heating to relatively high temperatures, of 180 ˚C, and thus have limited use.[3] Many Wolff rearrangement products are ring-strained and are susceptible to ring-open under high temperatures. In addition, SN2 substitution of the diazo group at the α-carbon can take place at lower temperatures than rearrangement, which results in byproducts. The greatest use of thermal Wolff rearrangements is the formation of carboxylic acid analogs, by interception of the ketene with high boiling solvents, such as aniline and phenol.[3]
Transition metals greatly lower the temperature of Wolff rearrangements, by stabilization of a metal-carbene intermediate. However, these carbenes can be so stable, as to not undergo rearrangement. Carbenes of rhodium, copper, and palladium are too stable and give non-Wolff products (primarily carbene insertion products).[2] The most commonly used metal catalyst is silver(I) oxide, although silver benzoate is also common. These reactions are generally run in the presence of a weak base, such as sodium carbonate or tertiary amines.[2]
Whereas thermal and metal mediated Wolff rearrangements date back to 1902,[3] photolytic methods are somewhat newer, with the first example of a photolytic Wolff rearrangement reported in 1951.[4] α-diazo ketones have two absorption bands, an allowed π→π* transition at 240–270 nm, and a formally forbidden π→σ* transition at 270–310 nm.[4] Medium or low-pressure mercury arc lamps can excite these respective transitions. Triplet sensitizers result in non-Wolff carbene byproducts, and thus are not useful in synthetic applications of the Wolff rearrangement.[2] However, they have been used to probe the mechanism of the Wolff rearrangement.
Synthetic uses
The Wolff rearrangement has a few retrons, depending on the reaction out of the ketene intermediate. A carboxylic acid derivative with an α-methylene group is a retron for an Arndt-Eistert type homologation. An acid in which the α-carbon belongs to a ring is a retron for a Wolff rearrangement ring contraction.
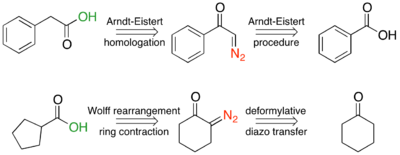
Homologation reactions
In the Arndt-Eistert homologation reaction, a carboxylic acid and thionyl chloride are reacted to generate an acid chloride. The acid chloride then reacts with diazomethane (R2 = H), or occasionally a diazoalkyl, via the Arndt-Eistert procedure, to generate an α-diazo ketone, which will undergo a metal-catalyzed or photolyzed Wolff rearrangement, to give a ketene. The ketene can be trapped with any weak acid, such as an alcohol or amine, to form the ester or amide. However, trapping with water, to form the acid is the most common form.
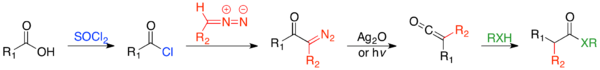
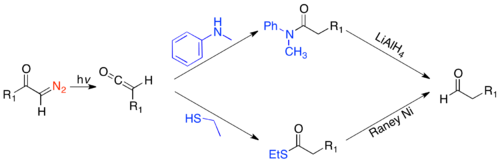
In the most basic form, where R2= H, RXH=H2O, the reaction lengthens the alkyl chain of a carboxylic acid by a methylene. However, there is great synthetic utility in the variety of reactions one can carry out, by varying the diazoalkyl and weak acid. The migrating group, R1 migrates with complete retention.[2] A very useful application of the Arndt-Eistert homologation forms the homologated aldehyde by either trapping the ketene with N-methyl aniline and reducing with lithium aluminum hydride, or trapping the ketene with ethanethiol and reducing with Raney nickel.[25][26]
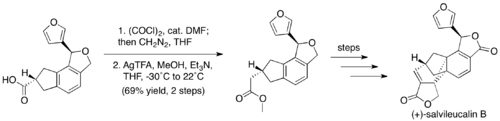
There exist many hundreds of examples of the Arndt-Eistert homologation in the literature.[27] Prominent examples in natural product total synthesis include the syntheses of (−)-indolizidine and (+)-macbecin.[28][29] A recent example of the Arndt-Eistert homologation is a step in the middle stage of Sarah Reisman's synthesis of (+)-salvileucalin B.[30]
Ring contractions
If the reactant is a cyclic α-diazo ketone, then Wolff-rearrangement products will be the one-carbon ring-contracted product. These reactions are generally concerted due to the s-cis conformation, and are photocatalyzed. The reaction below shows the concerted mechanism for the ring contraction of α-diazocyclohexanone, followed by trapping of the ketene with a weakly acidic nucleophile.
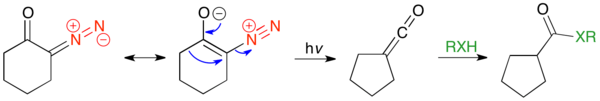
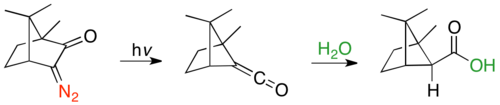
The first known example is the ring contracted Wolff rearrangement product of α-diazocamphor, and subsequent kinetic hydration of the ketene from the more sterically accessible "endo" face, to give exo-1,5,5-trimethylbicyclo[2.1.1]hexane-6-carboxylic acid.[31]
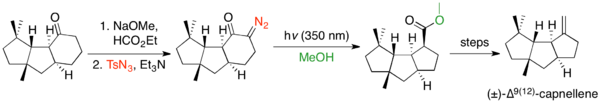
Ring contractions have been used extensively to build strained ring systems, as ring size does not impede the Wolff rearrangement, but often impedes other reactions. There are many examples where the Wolff rearrangement is used to contract cyclopentanone to cyclobutane.[32] The rearrangement is commonly used to form strained bicyclic and ring-fused systems. There exist a handful of examples of ring contractions from cyclobutanones to cyclopropanes.[33] The Wolff rearrangement is capable of contracting cyclohexanones to cyclopentanes, but is infrequently used to do so, because the Favorskii rearrangement accomplishes this transformation and the Wolff precursor is often more challenging to synthesize.[2] However, an example of a cyclohexanone ring contraction using deformylative diazo transfer, followed by a Wolff rearrangement, is Keiichiro Fukumoto's synthesis of (±)-∆9(12)-capnellene.[34]
Cycloaddition reactions
Ketene intermediates produced via the Wolff rearrangement are well known to undergo [2 + 2] thermal cycloadditions with olefins to form four-membered rings in both intermolecular and intramolecular reactions, examples of both are shown below.[35][36][37] Ketenes are able to undergo what is normally considered a forbidden [2 + 2] cycloaddition reaction because the ketene acts in an antarrafactial manner, leading to the Woodward-Hoffmann allowed [πs2 + πa2] cycloaddition.[36] Ketene [2 + 2] cycloadditions can be difficult reactions and give poor yields due to competing processes. The high energy aldoketene is very reactive and will cyclize with the diazo ketone starting material to produce butenolides and pyrazoles.[2]
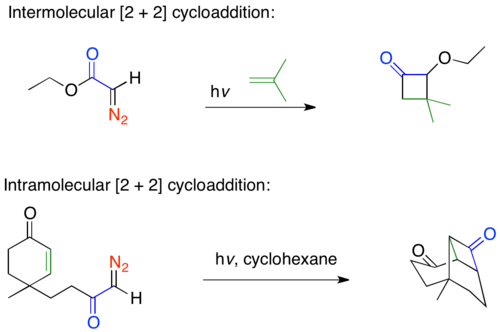
Ketene [2 + 2] cycloaddition reactions have been used in many total syntheses since Corey's use of the [2 + 2] cyclization in synthesizing the prostaglandins.[35] Robert Ireland's synthesis of (±)-aphidicolin uses the Wolff rearrangement to do a tandem ring-contraction, and [2 + 2] cycloaddition.[38]
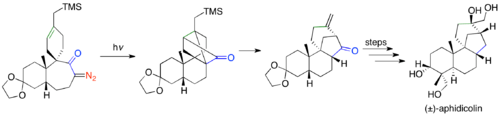
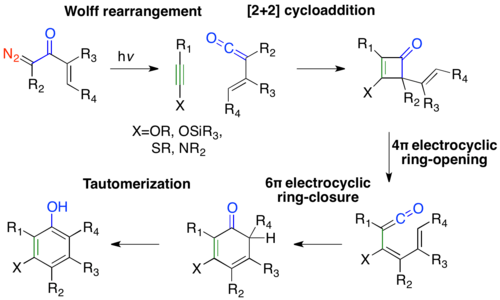
The Danheiser benzannulation photolyses α-diazo ketones and traps with an alkyne, which undergoes a pericyclic cascade, to ultimately form versatilely substituted phenols.[39] The first step in the benzannulation is the photolysis of an α-diazo ketone to form a vinylketene. The vinylketene then undergoes a [2 + 2] cycloaddition with an alkyne to form a 2-vinylcyclobutenone, which does a 4π electrocyclic ring-opening to generate a dienylketene. The dienylketene subsequently undergoes a 6π electrocyclic ring-closure followed by tautomerization, to form the phenolic benzannulated product.
Vinylogous Wolff rearrangements
The vinylogous Wolff rearrangement consists of a β,γ-unsaturated diazo ketone undergoing a Wolff rearrangement, and a formal 1,3-shift of the CH2CO2R group. The vinylogous Wolff rearrangement yields a γ,δ-unsaturated carboxylic acid derivative, which is the same retron as for the Claisen rearrangement. The variant was discovered when it was noticed that thermolysis of 1-diazo-3,3,3-triarylpropan-2-ones gave unexpected isomeric products.[40]

Copper (II) and rhodium (II) salts tend to give vinylogous Wolff rearranged products, and CuSO4 and Rh2(OAc)4 are the most commonly used catalysts.[41] This is because they promote metal carbene formation, which can add to the olefin to form a cyclopropane, which can reopen via a retro [2 + 2] to form a formally 1,3-shifted ketene (vis-à-vis a normal Wolff rearranged ketene), which can be trapped by a nucleophile to give the vinylogous Wolff product.[42]

See also
- Diazo
- Ketene
- Arndt-Eistert reaction
- Curtius rearrangement
- Schmidt reaction
References
- Kirmse, W. (2002). "100 Years of the Wolff Rearrangement". Eur. J. Org. Chem. 2002 (14): 2193. doi:10.1002/1099-0690(200207)2002:14<2193::AID-EJOC2193>3.0.CO;2-D.
- Gill, G. B. (1991) “The Wolff Rearrangement.” in Trost, B. M. Flemming, I. (eds.) Comp. Org. Synth. Oxford: Pergamon. 3:887. doi:10.1016/B978-0-08-052349-1.00085-8. ISBN 978-0-08-052349-1
- Wolff, L. (1902). "Ueber Diazoanhydride". Justus Liebigs Ann. Chem. 325 (2): 129–195. doi:10.1002/jlac.19023250202.
- Horner, L. Spietschka, E. Gross, A. W. (1951). "Zur Kenntnis der Umlagerungsvorgänge bei Diazo-ketonen, o-Chinondiaziden und Säureaziden". Justus Liebigs Ann. Chem. 573: 17–30. doi:10.1002/jlac.19515730103.CS1 maint: multiple names: authors list (link)
- Schroeter, G. (1909). "Über die Hofmann-Curtiussche, die Beckmannsche und die Benzilsäure-Umlagerung". Chem. Ber. 42 (2): 2336–2349. doi:10.1002/cber.190904202131.
- Pecile, C. Foffani, F. Chersetti, S. (1964). "The interaction of diazocarbonyl compounds with hydroxylic solvents". Tetrahedron. 20 (4): 823–829. doi:10.1016/S0040-4020(01)98414-5.CS1 maint: multiple names: authors list (link)
- Kaplan, F. Meloy, G. K. (1966). "The Structure of Diazoketones. A Study of Hindered Internal Rotation1,2". J. Am. Chem. Soc. 88 (5): 950–956. doi:10.1021/ja00957a017.CS1 maint: multiple names: authors list (link)
- Roth, H. D. Manion, M.L. (1976). "Solution photochemistry of diazoacetone. Wolff rearrangement and acetylmethylene". J. Am. Chem. Soc. 98 (11): 3392–3393. doi:10.1021/ja00427a067.CS1 maint: multiple names: authors list (link)
- Tomioka, H. Okuno, H. Kondo, S. Izawa, Y. (1980). "Direct evidence for ketocarbene-ketocarbene interconversion". J. Am. Chem. Soc. 102 (23): 7123–7125. doi:10.1021/ja00543a050.CS1 maint: multiple names: authors list (link)
- Regitz, M. W, Bartz. (1970). "Untersuchungen an Diazoverbindungen, VII. Vergleichende kinetische Untersuchungen zur thermischen Stabilität aliphatischer Diazoverbindungen". Chem. Ber. 103 (5): 1477–1485. doi:10.1002/cber.19701030519.CS1 maint: multiple names: authors list (link)
- Zeller, K. P. (1977). "Zur formylcarben-oxiren-isomerisierung". Tetrahedron Letters. 18 (8): 707–708. doi:10.1016/S0040-4039(01)92732-7.
- Zeller, K. P. Meier, H. Kolshorn, H. Mueller, E. (1972). "Zum Mechanismus der Wolff-Umlagerung". Chem. Ber. 105 (6): 1875–1886. doi:10.1002/cber.19721050610.CS1 maint: multiple names: authors list (link)
- Timm, U. Zeller, K. P. Meier, H. (1977). "Photolyse von 2-oxo-[2-13c]-1-diazocyclohexan. Ein beitrag zum oxiren-problem". Tetrahedron. 33 (4): 453–455. doi:10.1016/0040-4020(77)80104-X.CS1 maint: multiple names: authors list (link)
- Fenwick, J. Frater, G. Ogi, K. Strausz, O.P. (1973). "Mechanism of the Wolff rearrangement. IV. Role of oxirene in the photolysis of .alpha.-diazo ketones and ketenes". J. Am. Chem. Soc. 95: 124–132. doi:10.1021/ja00782a021.CS1 maint: multiple names: authors list (link)
- Zeller, K. P. Meier, H. Müller, E. (1972). "Untersuchungen zur Wolff-Umlagerung—II". Tetrahedron. 28 (23): 5831–5838. doi:10.1016/S0040-4020(01)88926-2.CS1 maint: multiple names: authors list (link)
- Wilds, A. L. Meader, A. L. (1948). "The use of higher diazohydrocarbons in the Arndt-Eistert synthesis". J. Org. Chem. 13 (5): 763–79. doi:10.1021/jo01163a024. PMID 18884425.CS1 maint: multiple names: authors list (link)
- Gallucci, R. R. Jones, M. Jr. (1985). "Photolysis of methyl 3-diazo-2-oxopropionate. Wolff migration of the carbomethoxy group". J. Org. Chem. 50 (22): 4404–4405. doi:10.1021/jo00222a047.CS1 maint: multiple names: authors list (link)
- Weygand, F. Dworschak, H. Koch, K. Konstas, S. (1961). "Reaktionen des Trifluoracetyl-carbäthoxy-carbens II. Mitteilung". Angew. Chem. 73 (11): 409. doi:10.1002/ange.19610731116.CS1 maint: multiple names: authors list (link)
- Arndt, F. Eistert, B. Partale, W. (1927). "Diazo-methan undo-Nitroverbindungen, II.:N-Oxy-isatin auso-Nitro-benzoylchlorid". Chem. Ber. 60 (6): 1364–1370. doi:10.1002/cber.19270600616.CS1 maint: multiple names: authors list (link)
- Franzen, V. (1957). "Eine neue Methode zur Darstellung α,β-ungesättiger Ketone. Zerfall der Diazoketone R—CO—CN2—CH2—R′". Justus Liebigs Annalen der Chemie. 602: 199. doi:10.1002/jlac.19576020116.
- Dakin, H. D. West, R. (1928). "A General Reaction of Amino Acids". J. Biol. Chem. 78: 91.CS1 maint: multiple names: authors list (link)
- Regitz, M. Liedhegener, A. (1966). "Reaktionen aktiver Methylenverbindungen mit Aziden, XII. Synthese von Diacyl-diazomethanen durch Diazogruppen-Übertragung". Chem. Ber. 99 (10): 3128–3147. doi:10.1002/cber.19660991010.CS1 maint: multiple names: authors list (link)
- Regitz, M. Rüter (1968). "Reaktionen CH-aktiver Verbindungen mit Aziden, XVIII. Synthese von 2-Oxo-1-diazo-cycloalkanen durch entformylierende Diazogruppen-Übertragung". J. Chem. Ber. 101 (4): 1263–1270. doi:10.1002/cber.19681010419.
- Danheiser, R. L.; Miller, R. F.; Brisbois, R. G.; Park, S. Z. (1990). "An improved method for the synthesis of .alpha.-diazo ketones". J Org Chem. 55 (6): 1959. doi:10.1021/jo00293a053.
- Weygand, F. Bestmann, H. J. (1960). "Neuere präparative Methoden der organischen Chemie III. Synthesen unter Verwendung von Diazoketonen". Angew. Chem. 72 (16): 535–554. doi:10.1002/ange.19600721602.CS1 maint: multiple names: authors list (link)
- Weygand, F. Bestmann, H. J. (1959). "Homologe Aldehyde aus Carbonsäuren". Chem. Ber. 92 (3): 528–529. doi:10.1002/cber.19590920303.CS1 maint: multiple names: authors list (link)
- Tao, Y. McKervey, M. A. (1994). "Organic Synthesis with .alpha.-Diazo Carbonyl Compounds". Chem. Rev. 94 (4): 1091–1160. doi:10.1021/cr00028a010.CS1 maint: multiple names: authors list (link)
- Jefford, C. W. Tang, Q. Zaslona, A. (1991). "Short, enantiogenic syntheses of (−)-indolizidine 167B and (+)-monomorine". J. Am. Chem. Soc. 113 (9): 3513–3518. doi:10.1021/ja00009a043.CS1 maint: multiple names: authors list (link)
- Evans, D. A. Miller, S. J. Ennis, M. D. (1993). "Asymmetric synthesis of the benzoquinoid ansamycin antitumor antibiotics: Total synthesis of (+)-macbecin". J. Org. Chem. 58 (2): 471–485. doi:10.1021/jo00054a035.CS1 maint: multiple names: authors list (link)
- Levin, S. Nani, R. R. Reisman, S. E. (2011). "Enantioselective total synthesis of (+)-salvileucalin B" (PDF). J. Am. Chem. Soc. 133 (4): 774–6. doi:10.1021/ja110192b. PMID 21174417.CS1 maint: multiple names: authors list (link)
- Horner, L. Spietschka, E. (1955). "Über Lichtreaktionen IV1): Bicyclo-[1.1.2]-hexan-Derivate als Ergebnis der Umlagerung des Diazocamphers im Licht". Chem. Ber. 88 (7): 934–939. doi:10.1002/cber.19550880705.CS1 maint: multiple names: authors list (link)
- Lowe, G. Ridley, D. D. (1973). "Synthesis of ?-lactams by photolytic Wolff rearrangement". J. Chem. Soc., Chem. Commun. (10): 328–329. doi:10.1039/c39730000328.CS1 maint: multiple names: authors list (link)
- Ueda, K. Toda, F. (1975). "Wolff Rearrangement of 2-Diazo-3,4-Bis(Diphenylmethylene)Cyclobutmone into 1,2-Bis (Diphenylmethylene) Cyclopropmes". Chem. Lett. 4 (7): 779–780. doi:10.1246/cl.1975.779.CS1 maint: multiple names: authors list (link)
- Ihara, M. Suzuki, T. Katogi, M. Taniguchi, N. Fukumoto, K. (1991). "A stereoselective total synthesis of (±)-Δ9(12)-capnellene via the intramolecular Diels–Alder approach". J. Chem. Soc. Chem. Commun. (9): 646–647. doi:10.1039/c39910000646.CS1 maint: multiple names: authors list (link)
- Corey, E. J. Arnold, Z. Hutton, J. (1970). "Total synthesis of prostaglandins E2 and F2α () via a tricarbocyclic intermediate". Tetrahedron Lett. 11 (4): 307–310. doi:10.1016/S0040-4039(00)61815-4. PMID 5414677.CS1 maint: multiple names: authors list (link)
- DoMinh, T. Strausz, O. P. (1970). "Cycloaddition of ethoxyketene to olefins". J. Am. Chem. Soc. 92 (6): 1766–1768. doi:10.1021/ja00709a062.CS1 maint: multiple names: authors list (link)
- Becker, D. Birnbaum, D. (1980). "Intramolecular photoaddition of ketenes to conjugated cycloalkenones". J. Org. Chem. 45 (4): 570–578. doi:10.1021/jo01292a004.CS1 maint: multiple names: authors list (link)
- Ireland, R. E. Dow, W. C. Godfrey, J. D. Thaisrivongs, S. (1984). "Total synthesis of (.+-.)-aphidicolin and (.+-.)-.beta.-chamigrene". J. Org. Chem. 49 (6): 1001–1013. doi:10.1021/jo00180a010.CS1 maint: multiple names: authors list (link)
- Danheiser, R. L. Brisbois, R. G. Kowalczyk, J. J. Miller, R. F. (1990). "An annulation method for the synthesis of highly substituted polycyclic aromatic and heteroaromatic compounds". J. Am. Chem. Soc. 112 (8): 3093–3100. doi:10.1021/ja00164a033.CS1 maint: multiple names: authors list (link)
- Wilds, A. L. van den Berghe, J. Winestock, C. H. von Trebra, R. L. Woolsey, N.F. (1962). "Abnormal Acids from the Arndt-Eistert Synthesis". J. Am. Chem. Soc. 84 (8): 1503–1504. doi:10.1021/ja00867a044.CS1 maint: multiple names: authors list (link)
- Smith, A. B. III, Toder, B. H., Branca, S. J. (1984). "Vinylogous Wolff rearrangement. 4. General reaction of .beta.,.gamma.-unsaturated .alpha.'-diazo ketones". J. Am. Chem. Soc. 106 (14): 3995–4001. doi:10.1021/ja00326a018.CS1 maint: multiple names: authors list (link)
- Zimmerman, H. E. Little, R. D. (1974). "Mechanistic and exploratory organic photochemistry. LXXXVII. Photochemical rearrangement of 4-aryl-substituted cyclopentenones. Low-temperature photochemistry and direct observation of reaction intermediates". J. Am. Chem. Soc. 96 (14): 4623–4630. doi:10.1021/ja00821a044.CS1 maint: multiple names: authors list (link)