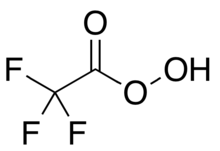
Trifluoroperacetic acid
Trifluoroperacetic acid (trifluoroperoxyacetic acid, TFPAA) is an organofluorine compound, the peroxy acid analog of trifluoroacetic acid, with the condensed structural formula CF
3COOOH.[Note 1] It is a strong oxidizing agent for organic oxidation reactions, such as in Baeyer–Villiger oxidations of ketones.[1] It is the most reactive of the organic peroxy acids, allowing it to successfully oxidise relatively unreactive alkenes to epoxides where other peroxy acids are ineffective.[2] It can also oxidise the chalcogens in some functional groups, such as by transforming selenoethers to selones.[3] It is a potentially explosive material[4] and is not commercially available, but it can be quickly prepared as needed.[5] Its use as a laboratory reagent was pioneered and developed by William D. Emmons.[6][7]
 | |
| Names | |
|---|---|
| IUPAC name
2,2,2-trifluoroethaneperoxoic acid | |
Other names
| |
| Identifiers | |
3D model (JSmol) |
|
| ChemSpider | |
PubChem CID |
|
| |
| |
| Properties | |
| C2HF3O3 | |
| Molar mass | 130.022 g·mol−1 |
| Appearance | colourless liquid |
| Boiling point | 162 °C (324 °F; 435 K) |
| Solubility | soluble in acetonitrile, dichloromethane, diethyl ether, sulfolane |
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |
| Infobox references | |
Properties
At standard ambient temperature and pressure, trifluoroperacetic acid is a colourless liquid with a boiling point of 162 °C.[8] It is soluble in acetonitrile, dichloromethane, diethyl ether, and sulfolane, and readily reacts with water.[5] Like all peroxy acids, it is potentially explosive and requires careful handling.[4] It is not commercially available, but can be made in the lab and stored for up to several weeks at −20 °C.[5] Some preparative methods result in mixtures containing residual hydrogen peroxide and trifluoroacetic acid, and heating such a mixture is extremely hazardous; the hydrogen peroxide can be decomposed using manganese dioxide for safety before heating.[5][8]
Preparation
Trifluoroperacetic acid can be easily prepared by an Organic Syntheses[9] process of treating trifluoroacetic anhydride with a concentrated (90%)[2] aqueous solution of hydrogen peroxide:
- CF
3COOCOCF
3 + H
2O
2 → CF
3COOOH + CF
3COOH
As the anhydride will form trifluoroacetic acid in contact with water, an excess of the anhydride also serves to remove the solvent from the peroxide reactant:[9]
- CF
3COOCOCF
3 + H
2O → 2 CF
3COOH
A more dilute hydrogen peroxide solution (30%) can be used to form trifluoroperacetic acid for some reactions from trifluoroacetic acid.[2]
- CF
3COOH + H
2O
2 → CF
3COOOH + H
2O
In order to avoid the danger of handling pure or highly concentrated solutions of hydrogen peroxide, hydrogen peroxide – urea can be used to give the peracid.[5] This method involves no water, so it gives a completely anhydrous peracid,[10] which is an advantage when the presence of water leads to side reactions during certain oxidation reactions.[11]
- CF
3COOCOCF
3 + H
2O
2·CO(NH
2)
2 → CF
3COOOH + CF
3COOH + CO(NH
2)
2
In cases where a pH buffering agent is needed for a synthesis and where the presence of water is tolerated, another approach has been developed. Reacting trifluoroacetic anhydride with sodium percarbonate, 2Na
2CO
3·3H
2O
2, yields trifluoroperacetic acid and sodium carbonate, obviating the need for an additional buffer.[5][12]
- 3 CF
3COOCOCF
3 + 4 Na
2CO
3·1 1⁄2H
2O
2 → 6 CF
3COOOH + 4 Na
2CO
3 + 3 H
2O
Trifluoroperacetic acid can also be generated in situ,[13] allowing it to react promptly with the target substrate rather than pre-synthesizing a batch of the reagent for later use.
Uses
iodo)benzene-3D-balls.png)
6H
5I(OOCCF
3)
2
Trifluoroperacetic acid is primarily used as an oxidising agent.[5][7] In September 1953, the Journal of the American Chemical Society published work by William D. Emmons and Arthur F. Ferris reporting that this reagent, generated in situ, was capable of oxidising aniline to nitrobenzene.[13] Over the following two years, Emmons reported a preparative method for this reagent and published six further manuscripts in this journal on its applications;[14][15][16] Emmons is remembered in part as the pioneer[6] and developer[7] of trifluoroperacetic acid as a laboratory reagent, which has since become useful as a reagent for many different types of synthetic reactions.
One example is the formation of the hypervalent iodine compound (bis(trifluoroacetoxy)iodo)benzene, (CF
3COO)
2IC
6H
5 which is used to carry out the Hofmann rearrangement under acidic conditions.[17] The hypervalent compound is accessible in two ways, and which is chosen usually depends on what materials are available: it can be prepared from its acetate analogue by an exchange reaction,[18] or by reacting iodobenzene with a combination of trifluoroperacetic acid and trifluoroacetic acid:[17]

Baeyer–Villiger oxidation
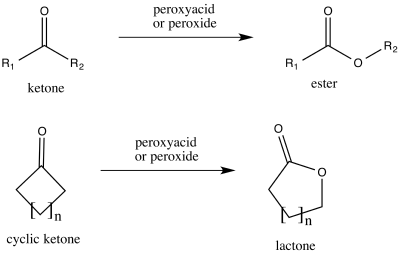
Trifluoroperacetic acid is one of the strongest reagents used for Baeyer–Villiger oxidations, as a consequence of its high acidity relative to similar peracids and peroxides.[19]:17 This reaction converts ketones to either straight-chain esters or lactones, and is named for Adolf von Baeyer and Victor Villiger, who first reported it 1899.[1] The reaction is believed to proceed via a Criegee intermediate[5] and demonstrates good regioselectivity and chemoselectivity for the position of oxygen atom insertion, along with retention of stereochemistry at the adjacent position, as can be seen in the following example. The disodium phosphate (Na
2HPO
4) is added as a pH buffer[2] to prevent the highly acidic trifluoroacetic acid byproduct from causing hydrolysis[20] or transesterification[21] of the ester product.
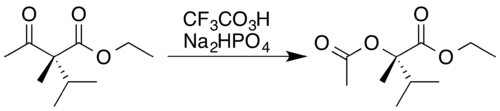
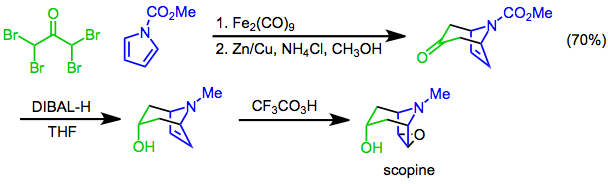
Epoxidation
The Prilezhaev reaction involves the conversion of an alkene to an epoxide using a peracid as the oxidant[22] and was first reported in 1909.[23] The reaction has been used as the final step of the synthesis of scopine, a tropane alkaloid. In this approach, a [4+3] cycloaddition mediated by diiron nonacarbonyl is used to construct the bicyclic skeleton, the hydroxyl functional group is then introduced by diastereoselective reduction of the ketone with diisobutylaluminum hydride, and the preparation completed with a Prilezhaev trifluoroperacetic acid epoxidation.[24]
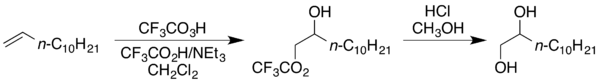
The high reactivity of trifluoroperacetic acid relative to other peroxy acids allows it to successfully oxidize relatively electron-poor alkenes such as 1-hexene and α,β-unsaturated esters such as methyl methacrylate, substrates that are generally resistant to peroxy-acid epoxidation.[2] Including additional buffered trifluoroacetic acid in the mixture gives a vicinal hydroxy–trifluoroacetate structure instead of an epoxide, which can be converted to the diol by treatment with acidic methanol, such as in the following conversion of 1-dodecene to 1,2-dodecanediol.[2]
In the case of an allyl alcohol compound with a proximate carbonyl functional group, the epoxide can undergo a ring-expansion reaction to form a dioxolane.[5][11] The process below was used as part of the total synthesis of neosporol, a natural product:[11][25]
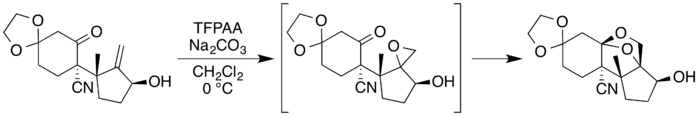
The preparation of the isomeric compound sporol involved a similar dioxolane formation. In this case, the use of trifluoroperacetic acid derived from hydrogen peroxide, which therefore presumably contained traces of water, gave mostly a hemiacetal rather than the closed-ring dioxolane. The use of the urea complex, which gave a water-free material, successfully gave the dioxolane as the major product.[11] The dioxolane is expanded to the 1,3-dioxane system found in sporol at a later step in the synthesis.[25]
Heteroatom oxidation
Functional groups containing heteroatoms in low oxidation states can be oxidised by trifluoroperacetic acid.[5][7] Common cases include the oxidation of iodine (for example, the formation of the hypervalent iodine compound from iodobenzene mentioned earlier), nitrogen, sulfur, and selenium.
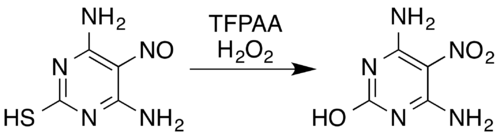
In the case of nitrogen-containing compounds, known transformations include oximes[5] and aromatic primary amines[15] to nitro compounds[7] (even with electron-withdrawing substituents, for example, pentafluoroaniline to pentafluoronitrobenzene[26]), nitrosamines to nitramines,[7][14] formation of aromatic N-oxides and aromatic azine N-oxides,[5][27] and conversion of nitroso compounds to nitro compounds or nitramines.[5] For example, a mixture of hydrogen peroxide and trifluoroperacetic acid oxidises the nitroso-substituted pyrimidine 4,6-diamino-5-nitrosopyrimidine-2-thiol to its nitro analogue while also removing the thiol moiety by oxidative hydrolytic desulfurization:[5][28]
In the case of chalcogen elements, sulfide moieties (R–S–R) can be oxidised by trifluoroperacetic acid to sulfoxide (R–S(O)–R) and/or sulfone (R–S(O)2–R) forms, depending on the conditions used.[5] In the analogous selenium system, trifluoroperacetic acid oxidation of selenoethers (R–Se–R) produces selones (R–Se(O)2–R) without the formation of the related selenoxides (R–Se(O)–R) as an isolable product,[3] a reaction which is particularly effective when the R is an aryl group.[29] A general approach to the formation of sulfinyl chlorides (RS(O)Cl) is the reaction of the corresponding thiol with sulfuryl chloride (SO
2Cl
2). In cases where the sulfenyl chloride (RSCl) results instead, a subsequent trifluoroperacetic acid oxidation affords the desired product, as in the case of 2,2,2-trifluoro-1,1-diphenylethanethiol:[30]
Cl.png)
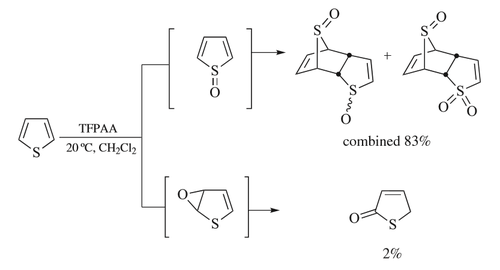
The trifluoroperacetic acid oxidation of thiophene illustrates competing pathways for reaction, with both S-oxidation and epoxidation being possible.[31][Note 2] The major pathway initially forms the sulfoxide, but this chemical promptly undergoes a Diels-Alder-type dimerisation before any further oxidation occurs—neither thiophene-S-oxide or thiophene-S,S-dioxide are found among the products of the reaction.[5][31] The dimer can then be oxidized further, converting one of the S-oxide moieties to an S,S-dioxide. In the minor reaction pathway, a Prilezhaev epoxidation[22] results in the formation of thiophene-2,3-epoxide that rapidly rearranges to the isomer thiophene-2-one.[31] Trapping experiments[35] demonstrate that this epoxide pathway is not an alternative reaction of the S-oxide intermediate, and isotopic labeling experiments demonstrate that a 1,2-hydride shift (an NIH shift) occurs and thus that a cationic intermediate is involved.[31] The choice of trifluoroperacetic acid preparation method is important as water suppresses the minor reaction pathway, likely because it acts as a competing base.[31]
Oxidation with acidic rearrangement
The use of trifluoroperacetic acid with boron trifluoride causes oxidation of alkenes and aromatic rings with concomitant rearrangement of the molecular skeleton.[5]
For alkenes, the reaction gives a ketone product, though the mechanistic process is not simply epoxidation followed by a BF3-catalyzed Wagner–Meerwein rearrangement:[36]
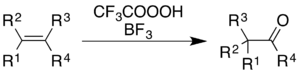
For aromatics, an example demonstrated in an Organic Syntheses report is the conversion of 1,2,3,4,5,6-hexamethylbenzene to 2,3,4,5,6,6-hexamethyl-2,4-cyclohexadienone:[9]
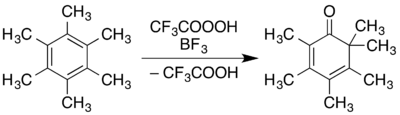
Oxidative cleavage of arenes
In addition to simple oxidation of aromatic rings to form carbonyl compounds (see § Oxidation with acidic rearrangement), trifluoroperacetic acid can fully cleave the carbon–carbon bonds within the ring. Unlike other oxidations of alkylaromatic structures, which yield benzoic acids and related compounds by cleavage of the alkyl chain at the reactive benzylic position, trifluoroperacetic acid causes an "inverse oxidation", cleaving the aromatic ring itself while leaving the alkyl group intact.[37][38]
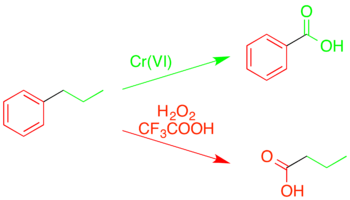
This selectivity for certain types of bonds allows it to be used to decompose complex mixtures of hydrocarbonds, such as coal, in order to determine structural details.[39][37]
Aromatic systems containing heteroatoms are resistant to this ring-opening as heteroatom oxidation occurs preferentially and deactivates the ring towards electrophilic attack by the peroxy acid. For example, purines, pyridines, and quinolines instead form N-oxides,[5] while sulfur systems like octafluorodibenzothiophene are converted to sulfones.[7][40]
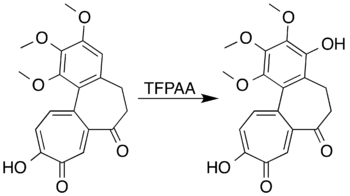
Aromatic systems with ring-activating substituents can be oxidised to form phenols instead of undergoing a ring-opening reaction. Mesitylene, for example, reacts with trifluoroperacetic acid to form mesitol (2,4,6-trimethylphenol).[7] Researchers attempting to form a lactone by Baeyer–Villiger oxidation of 7-oxodeacetamidocolchicine were unable to prepare the desired product, but did achieve oxidation of the aromatic ring to produce a phenol-derivative in high yield:[5][41]
Notes
- Three condensed structural formulae are used to represent trifluoroperacetic acid, CF
3COOOH, CF
3CO
3H, and CF
3C(O)OOH. They are equivalent and can be used interchangeably. - Such competitions can have biochemical significance. For example, it is known that the loop diuretic pharmaceutical agent tienilic acid acts as a suicide substrate at cytochrome P450 enzymes and that the process involves thiophene oxidation, but the oxidation pathway responsible remains unclear despite considerable research activity.[32][33][34]
References
- Kürti, László; Czakó, Barbara (2005). Strategic Applications of Named Reactions in Organic Synthesis. Elsevier Academic Press. p. 28. ISBN 9780124297852.
- Hiyama, Tamejiro (2000). "8.2 Trifluoroacetic acid and Trifluoroperacetic acid". Organofluorine Compounds: Chemistry and Applications. Springer Science & Business Media. pp. 255–257. ISBN 9783662041642.
- Kataoka, T.; Yoshimatsu, M. (1995). "Alkyl Chalcogenides: Selenium- and Tellurium-based Functional Groups". In Ley, Steven V. (ed.). Synthesis: Carbon with One Heteroatom Attached by a Single Bond. Comprehensive Organic Functional Group Transformations. Elsevier. pp. 277–296. ISBN 9780080423234.
- Carey, Francis A.; Sundberg, Richard J. (2007). "5.5 Addition Reactions Involving Epoxides". Advanced Organic Chemistry: Part A: Structure and Mechanisms (5th ed.). Springer Science & Business Media. pp. 503–514. ISBN 9780387448978.
- Caster, Kenneth C.; Rao, A. Somasekar; Mohan, H. Rama; McGrath, Nicholas A.; Brichacek, Matthew (2012). "Trifluoroperacetic Acid". Encyclopedia of Reagents for Organic Synthesis. e-EROS Encyclopedia of Reagents for Organic Synthesis. doi:10.1002/047084289X.rt254.pub2. ISBN 978-0471936237.
- Freeman, Jeremiah P. (November 14, 2002). "William D. Emmons: November 18, 1924 – December 8, 2001" (PDF). Org. Synth. 80: xxvii–xxix. Archived from the original (PDF) on March 16, 2015. Retrieved January 21, 2017.
- Chambers, Richard D. (2004). "Functional Compounds Containing Oxygen, Sulphur or Nitrogen and their Derivatives". Fluorine in Organic Chemistry. CRC Press. pp. 242–243. ISBN 9780849317903.
- Luxon, S. G. (1992). Hazards in the Chemical Laboratory (5th ed.). Royal Society of Chemistry. p. 627. ISBN 9780851862293.
- Hart, Harold; Lange, Richard M.; Collins, Peter M. (1968). "2,3,4,5,6,6-Hexamethyl-2,4-cyclohexadien-1-one". Organic Syntheses. 48: 87. doi:10.15227/orgsyn.048.0087.; Collective Volume, 5, p. 598
- Cooper, Mark S.; Heaney, Harry; Newbold, Amanda J.; Sanderson, William R. (1990). "Oxidation Reactions Using Urea–Hydrogen Peroxide; A Safe Alternative to Anhydrous Hydrogen Peroxide". Synlett. 1990 (9): 533–535. doi:10.1055/s-1990-21156.
- Ziegler, Fredrick E.; Metcalf, Chester A.; Nangia, Ashwini; Schulte, Gayle (1993). "Structure and total synthesis of sporol and neosporol". J. Am. Chem. Soc. 115 (7): 2581–2589. doi:10.1021/ja00060a006.
- Kang, Ho-Jung; Jeong, Hee-Sun (1996). "New Method of Generating Trifluoroperoxyacetic acid for the Baeyer-Villiger Reaction". Bull. Korean Chem. Soc. 17 (1): 5–6.
- Emmons, William D.; Ferris, Arthur F. (1953). "Oxidation Reactions with Pertrifluoroacetic Acid". J. Am. Chem. Soc. 75 (18): 4623–4624. doi:10.1021/ja01114a539.
- Emmons, William D. (1954). "Peroxytrifluoroacetic Acid. I. The Oxidation of Nitrosamines to Nitramines". J. Am. Chem. Soc. 76 (13): 3468–3470. doi:10.1021/ja01642a029.
- Emmons, William D. (1954). "Peroxytrifluoroacetic Acid. II. The Oxidation of Anilines to Nitrobenzenes". J. Am. Chem. Soc. 76 (13): 3470–3472. doi:10.1021/ja01642a030.
- Emmons, William D.; Pagano, Angelo S.; Freeman, Jeremiah P. (1954). "Peroxytrifluoroacetic Acid. III. The Hydroxylation of Olefins". J. Am. Chem. Soc. 76 (13): 3472–3474. doi:10.1021/ja01642a031.
Emmons, William D.; Pagano, Angelo S. (1955). "Peroxytrifluoroacetic Acid. IV. The Epoxidation of Olefins". J. Am. Chem. Soc. 77 (1): 89–92. doi:10.1021/ja01606a029.
Emmons, William D.; Lucas, George B. (1955). "Peroxytrifluoroacetic Acid. V. The Oxidation of Ketones to Esters". J. Am. Chem. Soc. 77 (8): 2287–2288. doi:10.1021/ja01613a077.
Emmons, William D.; Pagano, Angelo S. (1955). "Peroxytrifluoroacetic Acid. VI. The Oxidation of Oximes to Nitroparaffins". J. Am. Chem. Soc. 77 (17): 4557–4559. doi:10.1021/ja01622a036. - Aubé, Jeffrey; Fehl, Charlie; Liu, Ruzhang; McLeod, Michael C.; Motiwala, Hashim F. (1993). "6.15 Hofmann, Curtius, Schmidt, Lossen, and Related Reactions". Heteroatom Manipulations. Comprehensive Organic Synthesis II. 6. pp. 598–635. doi:10.1016/B978-0-08-097742-3.00623-6. ISBN 9780080977430.
- Almond, M. R.; Stimmel, J. B.; Thompson, E. A.; Loudon, G. M. (1988). "Hofmann Rearrangement Under Mildly Acidic Conditions Using [I,I-Bis(Trifluoroacetoxy)]Iodobenzene: Cyclobutylamine Hydrochloride from Cyclobutanecarboxamide". Organic Syntheses. 66: 132. doi:10.15227/orgsyn.066.0132.; Collective Volume, 8, p. 132
- Myers, Andrew G. "Chemistry 115 Handouts: Oxidation" (PDF). Harvard University. Retrieved 10 January 2017.
- Carruthers, William (1971). "6.3 Oxidation of Olefins". Some Modern Methods of Organic Synthesis. Cambridge University Press. pp. 259–280. ISBN 9780521096430.
- Carruthers, William (1971). "6.5 Baeyer–Villiger oxidation of ketones". Some Modern Methods of Organic Synthesis. Cambridge University Press. pp. 287–290. ISBN 9780521096430.
- Hagen, Timothy J. (2007). "Prilezhaev reaction". In Li, Jie Jack; Corey, E. J. (eds.). Name Reactions of Functional Group Transformations. John Wiley & Sons. pp. 274–281. ISBN 9780470176504.
- Prileschajew, Nikolaus (1909). "Oxydation ungesättigter Verbindungen mittels organischer Superoxyde" [Oxidation of unsaturated compounds by means of organic superoxides]. Ber. Dtsch. Chem. Ges. (in German). 42 (4): 4811–4815. doi:10.1002/cber.190904204100.
- Hayakawa, Y.; Baba, Y.; Makino, S.; Noyori, R. (1978). "Carbon-carbon bond formation promoted by transition metal carbonyls. 19. General synthesis of tropane alkaloids via the polybromo ketone-iron carbonyl reaction". J. Am. Chem. Soc. 100 (6): 1786–1791. doi:10.1021/ja00474a021.
- Pirrung, Michael C.; Morehead, Andrew T.; Young, Bruce G., eds. (2000). "10. Neosporol, Sporol". Part B: Bicyclic and Tricyclic Sesquiterpenes. The Total Synthesis of Natural Products. 11. John Wiley & Sons. pp. 222–224. ISBN 9780470129630.
- Brooke, G. M.; Burdon, J.; Tatlow, J. C. (1961). "Aromatic polyfluoro-compounds. Part VII. The reaction of pentafluoronitrobenzene with ammonia". J. Chem. Soc.: 802–807. doi:10.1039/JR9610000802.
- Williams, W. Michael; Dolbier, William R. (1969). "Thermal and photochemical rearrangements of azine oxides. I. Pyrolytic decomposition to nitriles". J. Org. Chem. 34 (1): 155–157. doi:10.1021/jo00838a034.
- Taylor, Edward C.; McKillop, Alexander (1965). "A New Synthesis of 5-Nitropyrimidines". J. Org. Chem. 30 (9): 3153–3155. doi:10.1021/jo01020a067.
- Taylor, P. C. (1995). "Vinyl and Aryl Chalcogenides: Sulfur-, Selenium- and Tellurium-based Functional Groups". In Ley, Steven V. (ed.). Synthesis: Carbon with One Heteroatom Attached by a Single Bond. Comprehensive Organic Functional Group Transformations. Elsevier. pp. 705–736. ISBN 9780080423234.
- Page, P. C. B.; Wilkes, R. D.; Reynolds, D. (1995). "Alkyl Chalcogenides: Sulfur-based Functional Groups". In Ley, Steven V. (ed.). Synthesis: Carbon with One Heteroatom Attached by a Single Bond. Comprehensive Organic Functional Group Transformations. Elsevier. pp. 113–276. ISBN 9780080423234.
- Treiber, Alexander (2002). "Mechanism of the Aromatic Hydroxylation of Thiophene by Acid-Catalyzed Peracid Oxidation". J. Org. Chem. 67 (21): 7261–7266. doi:10.1021/jo0202177. PMID 12375952.
- Mansuy, Daniel; Valadon, Philippe; Erdelmeier, Irene; López Garcia, Pilar; Amar, Claudine; Girault, Jean-Pierre; Dansette, Patrick M. (1991). "Thiophene S-Oxides as new Reactive Metabolites: Formation by Cytochrome P-450 Dependent Oxidation and Reaction with Nucleophiles". J. Am. Chem. Soc. 113 (20): 7825–7826. doi:10.1021/ja00020a089.
- Correia, Maria A.; Hollenberg, Paul F. (2015). "Inhibition of Cytochrome P450 Enzymes". In Ortiz de Montellano, Paul R. (ed.). Cytochrome P450: Structure, Mechanism, and Biochemistry (4th ed.). Springer. pp. 177–260. ISBN 9783319121086.
- Macherey, Anne-Christine; Dansette, Patrick M. (2015). "Biotransformations Leading to Toxic Metabolites:Chemical Aspects". In Wermuth, Camille Georges; Aldous, David; Raboisson, Pierre; Rognan, Didier (eds.). The Practice of Medicinal Chemistry (4th ed.). Elsevier. pp. 585–614. ISBN 9780124172135.
- Anslyn, Eric V.; Dougherty, Dennis A. (2006). "8.8 Miscellaneous Experiments for Studying Mechanism". Modern Physical Organic Chemistry. University Science Books. pp. 471–482. ISBN 9781891389313.
- Hart, Harold; Lerner, Lawrence R. (1967). "Oxidations with peroxytrifluoroacetic acid-boron trifluoride. IX. Direct oxidation of alkenes to ketones using peroxytrifluoroacetic acid–boron fluoride". J. Org. Chem. 32 (9): 2669–2673. doi:10.1021/jo01284a004.
- Deno, Norman C.; Greigger, Barbara A.; Stroud, Stephen G. (1978). "New method for elucidating the structures of coal". Fuel. 57 (8): 455–459. doi:10.1016/0016-2361(78)90153-9.
- Deno, Norman C.; Greigger, Barbara A.; Messer, Lauren A.; Meyer, Michael D.; Stroud, Stephen G. (1977). "Aromatic ring oxidation of alkylbenzenes". Tetrahedron Lett. 18 (20): 1703–1704. doi:10.1016/S0040-4039(01)93253-8.
- Deno, Norman C.; Curry, Kenneth W.; Greigger, Barbara A.; Jones, A. Daniel; Rakitsky, Walter G.; Smith, Karen A.; Wagner, Karen; Minard, Robert D. (1980). "Dihydroaromatic structure of Illinois No. 6 Monterey coal". Fuel. 59 (10): 694–698. doi:10.1016/0016-2361(80)90021-6.
- Chambers, R. D.; Cunningham, J. A.; Spring, D. J. (1968). "Polyfluoroaryl organometallic compounds. Part VIII. Synthesis of and nucleophilic substitution in octafluorodibenzofuran". J. Chem. Soc. C: 1560–1565. doi:10.1039/J39680001560.
- Berg, Ulf; Bladha, Håkan; Mpamposa, Konstantinos (2004). "Stereochemical variations on the colchicine motif. Peracid oxidation of thiocolchicone. Synthesis, conformation and inhibition of microtubule assembly". Org. Biomol. Chem. 2 (14): 2125–2130. doi:10.1039/B402840F. PMID 15254641.