Immune tolerance
Immune tolerance, or immunological tolerance, or immunotolerance, is a state of unresponsiveness of the immune system to substances or tissue that have the capacity to elicit an immune response in a given organism. It is induced by prior exposure to that specific antigen[1][2] and contrasts with conventional immune-mediated elimination of foreign antigens (see Immune response). Tolerance is classified into central tolerance or peripheral tolerance depending on where the state is originally induced—in the thymus and bone marrow (central) or in other tissues and lymph nodes (peripheral). The mechanisms by which these forms of tolerance are established are distinct, but the resulting effect is similar.
Immune tolerance is important for normal physiology. Central tolerance is the main way the immune system learns to discriminate self from non-self. Peripheral tolerance is key to preventing over-reactivity of the immune system to various environmental entities (allergens, gut microbes, etc.). Deficits in central or peripheral tolerance also cause autoimmune disease, resulting in syndromes such as systemic lupus erythematosus,[3] rheumatoid arthritis, type 1 diabetes,[4] autoimmune polyendocrine syndrome type 1 (APS-1),[5] and immunodysregulation polyendocrinopathy enteropathy X-linked syndrome (IPEX),[6] and potentially contribute to asthma, allergy,[7] and inflammatory bowel disease.[4] And immune tolerance in pregnancy is what allows a mother animal to gestate a genetically distinct offspring with an alloimmune response muted enough to prevent miscarriage.
Tolerance, however, also has its negative tradeoffs. It allows for some pathogenic microbes to successfully infect a host and avoid elimination.[8] In addition, inducing peripheral tolerance in the local microenvironment is a common survival strategy for a number of tumors that prevents their elimination by the host immune system.[9]
Historical background
The phenomenon of immune tolerance was first described by Ray D. Owens in 1945, who noted that dizygotic twin cattle sharing a common placenta also shared a stable mixture of each other's red blood cells (though not necessarily 50/50), and retained that mixture throughout life.[1] Although Owens did not use the term immune tolerance, his study showed the body could be tolerant of these foreign tissues. This observation was experimentally validated by Leslie Brent, Rupert E. Billingham and Peter Medawar in 1953, who showed by injecting foreign cells into fetal or neonatal mice, they could become accepting of future grafts from the same foreign donor. However, they were not thinking of the immunological consequences of their work at the time: as Medawar explains:
- "We did not set out with the idea in mind of studying the immunological consequences of the phenomenon described by Owen; on the contrary, we had been goaded by Dr. H.P. Donald into trying to devise a foolproof method of distinguishing monozygotic from dizygotic twins... ."[1]
However, these discoveries, and the host of allograft experiments and observations of twin chimerism they inspired, were seminal for the theories of immune tolerance formulated by Sir Frank McFarlane Burnet and Frank Fenner, who were the first to propose the deletion of self-reactive lymphocytes to establish tolerance, now termed clonal deletion.[10] Burnet and Medawar were ultimately credited for "the discovery of acquired immune tolerance" and shared the Nobel Prize in Physiology or Medicine in 1960.[1]
Definitions and usage
In their Nobel Lecture, Medawar and Burnet define immune tolerance as "a state of indifference or non-reactivity towards a substance that would normally be expected to excite an immunological response."[1] Other more recent definitions have remained more or less the same. The 8th edition of Janeway's Immunobiology defines tolerance as "immunologically unresponsive…to another's tissues.".[2]
Immune tolerance encompasses the range of physiological mechanisms by which the body reduces or eliminates an immune response to particular agents. It is used to describe the phenomenon underlying discrimination of self from non-self, suppressing allergic responses, allowing chronic infection instead of rejection and elimination, and preventing attack of fetuses by the maternal immune system. Typically, a change in the host, not the antigen, is implied.[1] Though some pathogens can evolve to become less virulent in host-pathogen coevolution,[11] tolerance does not refer to the change in the pathogen but can be used to describe the changes in host physiology. Immune tolerance also does not usually refer to artificially induced immunosuppression by corticosteroids, lymphotoxic chemotherapy agents, sublethal irradiation, etc. Nor does it refer to other types of non-reactivity such as immunological paralysis.[12] In the latter two cases, the host's physiology is handicapped but not fundamentally changed.
Immune tolerance is formally differentiated into central or peripheral;[2] however, alternative terms such as "natural" or "acquired" tolerance have at times been used to refer to establishment of tolerance by physiological means or by artificial, experimental, or pharmacological means.[13] These two methods of categorization are sometimes confused, but are not equivalent—central or peripheral tolerance may be present naturally or induced experimentally. This difference is important to keep in mind.
Central tolerance
Central tolerance refers to the tolerance established by deleting autoreactive lymphocyte clones before they develop into fully immunocompetent cells. It occurs during lymphocyte development in the thymus[14][15] and bone marrow for T and B lymphocytes, respectively. In these tissues, maturing lymphocytes are exposed to self-antigens presented by medullary thymic epithelial cells and thymic dendritic cells, or bone marrow cells. Self-antigens are present due to endogenous expression, importation of antigen from peripheral sites via circulating blood, and in the case of thymic stromal cells, expression of proteins of other non-thymic tissues by the action of the transcription factor AIRE.
Those lymphocytes that have receptors that bind strongly to self-antigens are removed by induction of apoptosis of the autoreactive cells, or by induction of anergy, a state of non-activity.[16] Weakly autoreactive B cells may also remain in a state of immunological ignorance where they simply do not respond to stimulation of their B cell receptor. Some weakly self-recognizing T cells are alternatively differentiated into natural regulatory T cells (nTreg cells), which act as sentinels in the periphery to calm down potential instances of T cell autoreactivity (see peripheral tolerance below).[2]
The deletion threshold is much more stringent for T cells than for B cells since T cells alone can cause direct tissue damage. Furthermore, it is more advantageous for the organism to let its B cells recognize a wider variety of antigen so it can produce antibodies against a greater diversity of pathogens. Since the B cells can only be fully activated after confirmation by more self-restricted T cells that recognize the same antigen, autoreactivity is held in check.[16]
This process of negative selection ensures that T and B cells that could initiate a potent immune response to the host's own tissues are eliminated while preserving the ability to recognize foreign antigens. It is the step in lymphocyte education that is key for preventing autoimmunity (entire process detailed here). Lymphocyte development and education is most active in fetal development but continues throughout life as immature lymphocytes are generated, slowing as the thymus degenerates and the bone marrow shrinks in adult life.
Peripheral tolerance
Peripheral tolerance develops after T and B cells mature and enter the peripheral tissues and lymph nodes.[2] It is established by a number of partly overlapping mechanisms that mostly involve control at the level of T cells, especially CD4+ helper T cells, which orchestrate immune responses and give B cells the confirmatory signals they need in order to produce antibodies. Inappropriate reactivity toward normal self-antigen that was not eliminated in the thymus can occur, since the T cells that leave the thymus are relatively but not completely safe. Some will have receptors (TCRs) that can respond to self-antigens that:
- are present in such high concentration outside the thymus that they can bind to "weak" receptors.
- the T cell did not encounter in the thymus (such as, tissue-specific molecules like those in the islets of Langerhans, brain, or spinal cord not expressed by AIRE in thymic tissues).
Those self-reactive T cells that escape intrathymic negative selection in the thymus can inflict cell injury unless they are deleted or effectively muzzled in the peripheral tissue chiefly by nTreg cells (see central tolerance above).
Appropriate reactivity toward certain antigens can also be quieted by induction of tolerance after repeated exposure, or exposure in a certain context. In these cases, there is a differentiation of naïve CD4+ helper T cells into induced Treg cells (iTreg cells) in the peripheral tissue or nearby lymphoid tissue (lymph nodes, mucosal-associated lymphoid tissue, etc.). This differentiation is mediated by IL-2 produced upon T cell activation, and TGF-β from any of a variety of sources, including tolerizing dendritic cells (DCs), other antigen presenting cells, or in certain conditions surrounding tissue.[8]
Treg cells are not the only cells that mediate peripheral tolerance. Other regulatory immune cells include T cell subsets similar to but phenotypically distinct from Treg cells, including TR1 cells that make IL-10 but do not express Foxp3, TGF-β-secreting TH3 cells, as well as other less well-characterized cells that help establish a local tolerogenic environment.[17] B cells also express CD22, a non-specific inhibitor receptor that dampens B cell receptor activation. A subset of B regulatory cells that makes IL-10 and TGF-β also exists.[18] Some DCs can make Indoleamine 2,3-dioxygenase (IDO) that depletes the amino acid tryptophan needed by T cells to proliferate and thus reduce responsiveness. DCs also have the capacity to directly induce anergy in T cells that recognize antigen expressed at high levels and thus presented at steady-state by DCs.[19] In addition, FasL expression by immune privileged tissues can result in activation-induced cell death of T cells.[20]
nTreg vs. iTreg cells
The involvement of T cells, later classified as Treg cells, in immune tolerance was recognized in 1995 when animal models showed that CD4+ CD25+ T cells were necessary and sufficient for the prevention of autoimmunity in mice and rats.[17] Initial observations showed removal of the thymus of a newborn mouse resulted in autoimmunity, which could be rescued by transplantation of CD4+ T cells. A more specific depletion and reconstitution experiment established the phenotype of these cells as CD4+ and CD25+. Later in 2003, experiments showed that Treg cells were characterized by the expression of the Foxp3 transcription factor, which is responsible for the suppressive phenotype of these cells.[17]
It was assumed that, since the presence of the Treg cells originally characterized was dependent on the neonatal thymus, these cells were thymically derived. By the mid-2000s, however, evidence was accruing of conversion of naïve CD4+ T cells to Treg cells outside of the thymus.[8] These were later defined as induced or iTreg cells to contrast them with thymus-derived nTreg cells. Both types of Treg cells quieten autoreactive T cell signaling and proliferation by cell-contact-dependent and -independent mechanisms including:[21]
- Contact-dependent:
- Granzyme or perforin secretion upon contact
- Upregulation of cAMP after contact, inducing anergy (reduced proliferation and IL-2 signaling)
- Interaction with B7 on T cells
- Downregulation of CD80/CD86 costimultory molecules on antigen presenting cells upon interaction with CTLA-4 or lymphocyte function-associated antigen 1 (LFA-1)
- Contact-independent
nTreg cells and iTreg cells, however, have a few important distinguishing characteristics that suggest they have different physiological roles:[8]
- nTreg cells develop in the thymus; iTreg cells develop outside the thymus in chronically inflamed tissue, lymph nodes, spleen, and gut-associated lymphoid tissue (GALT).
- nTreg cells develop from Foxp3- CD25+ CD4+ cells while iTreg cells develop from Foxp3+ CD25- CD4- cells (both become Foxp3+ CD25+CD4+).
- nTreg cells, when activated, require CD28 costimulation, while iTreg cells require CTLA-4 costimulation.
- nTreg cells are specific, modestly, for self-antigen while iTreg cells recognize allergens, commensal bacteria, tumor antigens, alloantigens, and self-antigens in inflamed tissue.
Tolerance in physiology and medicine
Allograft tolerance
Immune recognition of non-self-antigens typically complicates transplantation and engrafting of foreign tissue from an organism of the same species (allografts), resulting in graft reaction. However, there are two general cases in which an allograft may be accepted. One is when cells or tissue are grafted to an immune-privileged site that is sequestered from immune surveillance (like in the eye or testes) or has strong molecular signals in place to prevent dangerous inflammation (like in the brain). The second is when a state of tolerance has been induced, either by previous exposure to the antigen of the donor in a manner that causes immune tolerance rather than sensitization in the recipient, or after chronic rejection. Long-term exposure to a foreign antigen from fetal development or birth may result in establishment of central tolerance, as was observed in Medawar's mouse-allograft experiments.[1] In usual transplant cases, however, such early prior exposure is not possible. Nonetheless, a few patients can still develop allograft tolerance upon cessation of all exogenous immunosuppressive therapy, a condition referred to as operational tolerance.[22][23] CD4+ Foxp3+ Treg cells, as well as CD8+ CD28- regulatory T cells that dampen cytotoxic responses to grafted organs, are thought to play a role.[16] In addition, genes involved in NK cell and γδT cell function associated with tolerance have been implicated for liver transplant patients.[23] The unique gene signatures of these patients implies their physiology may be predisposed toward immune tolerance.
Fetal development
The fetus has a different genetic makeup than the mother, as it also translates its father's genes, and is thus perceived as foreign by the maternal immune system. Women who have borne multiple children by the same father typically have antibodies against the father's red blood cell and major histocompatibility complex (MHC) proteins.[2] However, the fetus usually is not rejected by the mother, making it essentially a physiologically tolerated allograft. It is thought that the placental tissues which interface with maternal tissues not only try to escape immunological recognition by downregulating identifying MHC proteins but also actively induce a marked peripheral tolerance. Placental trophoblast cells express a unique Human Leukocyte Antigen (HLA-G) that inhibits attack by maternal NK cells. These cells also express IDO, which represses maternal T cell responses by amino acid starvation. Maternal T cells specific for paternal antigens are also suppressed by tolerogenic DCs and activated iTregs or cross-reacting nTregs.[24] Some maternal Treg cells also release soluble fibrinogen-like proteins 2 (sFGL2), which suppresses the function of DCs and macrophages involved in inflammation and antigen presentation to reactive T cells[24] These mechanisms altogether establish an immune-privileged state in the placenta that protects the fetus. A break in this peripheral tolerance results in miscarriage and fetal loss.[25] (for more information, see Immune tolerance in pregnancy).
The microbiome
The skin and digestive tract of humans and many other organisms is colonized with an ecosystem of microorganisms that is referred to as the microbiome. Though in mammals a number of defenses exist to keep the microbiota at a safe distance, including a constant sampling and presentation of microbial antigens by local DCs, most organisms do not react against commensal microorganisms and tolerate their presence. Reactions are mounted, however, to pathogenic microbes and microbes that breach physiological barriers. Peripheral mucosal immune tolerance, in particular, mediated by iTreg cells and tolerogenic antigen-presenting cells, is thought to be responsible for this phenomenon. In particular, specialized gut CD103+ DCs that produce both TGF-β and retinoic acid efficiently promotes the differentiation of iTreg cells in the gut lymphoid tissue.[8] Foxp3- TR1 cells that make IL-10 are also enriched in the intestinal lining.[2] Break in this tolerance is thought to underlie the pathogenesis of inflammatory bowel diseases like Crohn's disease and ulcerative colitis.[4]
Oral tolerance and hypersensitivity
Oral tolerance refers to a specific type of peripheral tolerance induced by antigens given by mouth and exposed to the gut mucosa and its associated lymphoid tissues.[13] The hypo-responsiveness induced by oral exposure is systemic and can reduce hypersensitivity reactions in certain cases. Records from 1829 indicate that American Indians would reduce contact hypersensitivity from poison ivy by consuming leaves of related Rhus species; however, contemporary attempts to use oral tolerance to ameliorate autoimmune diseases like rheumatoid arthritis and other hypersensitivity reactions have been mixed.[13] The systemic effects of oral tolerance may be explained by the extensive recirculation of immune cells primed in one mucosal tissue in another mucosal tissue, allowing extension of mucosal immunity.[26] The same probably occurs for cells mediating mucosal immune tolerance.
Oral tolerance may depend on the same mechanisms of peripheral tolerance that limit inflammation to bacterial antigens in the microbiome since both involve the gut-associated lymphoid tissue. It may also have evolved to prevent hypersensitivity reactions to food proteins.[27] It is of immense immunological importance, since it is a continuous natural immunologic event driven by exogenous antigen.
Allergy and hypersensitivity reactions in general are traditionally thought of as misguided or excessive reactions by the immune system, possibly due to broken or underdeveloped mechanisms of peripheral tolerance. Usually, Treg cells, TR1, and Th3 cells at mucosal surfaces suppress type 2 CD4 helper cells, mast cells, and eosinophils, which mediate allergic response. Deficits in Treg cells or their localization to mucosa have been implicated in asthma and atopic dermatitis.[28] Attempts have been made to reduce hypersensitivity reactions by oral tolerance and other means of repeated exposure. Repeated administration of the allergen in slowly increasing doses, subcutaneously or sublingually appears to be effective for allergic rhinitis.[29] Repeated administration of antibiotics, which can form haptens to cause allergic reactions, can also reduce antibiotic allergies in children.[30]
The tumor microenvironment
Immune tolerance is an important means by which growing tumors, which have mutated proteins and altered antigen expression, prevent elimination by the host immune system. It is well recognized that tumors are a complex and dynamic population of cells composed of transformed cells as well as stromal cells, blood vessels, tissue macrophages, and other immune infiltrates.[9][31] These cells and their interactions all contribute to the changing tumor microenvironment, which the tumor largely manipulates to be immunotolerant so as to avoid elimination. There is an accumulation of metabolic enzymes that suppress T cell proliferation and activation, including IDO and arginase, and high expression of tolerance-inducing ligands like FasL, PD-1, CTLA-4, and B7.[9][20] Pharmacologic monoclonal antibodies targeted against some of these ligands has been effective in treating cancer.[32] Tumor-derived vesicles known as exosomes have also been implicated promoting differentiation of iTreg cells and myeloid derived suppressor cells (MDSCs), which also induce peripheral tolerance.[9][33] In addition to promoting immune tolerance, other aspects of the microenvironment aid in immune evasion and induction of tumor-promoting inflammation.
Evolution
Though the exact evolutionary rationale behind the development of immunological tolerance is not completely known, it is thought to allow organisms to adapt to antigenic stimuli that will consistently be present instead of expending considerable resources fighting it off repeatedly. Tolerance in general can be thought of as an alternative defense strategy that focuses on minimizing impact of an invader on host fitness, instead of on destroying and eliminating the invader.[34] Such efforts may have a prohibitive cost on host fitness. In plants, where the concept was originally used, tolerance is defined as a reaction norm of host fitness over a range of parasite burdens, and can be measured from the slope of the line fitting these data.[35] Immune tolerance may constitute one aspect of this defense strategy, though other types of tissue tolerance have been described.[34]
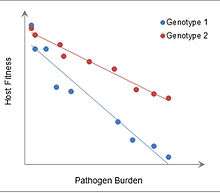
The advantages of immune tolerance, in particular, may be seen in experiments with mice infected with malaria, in which more tolerant mice have higher fitness at greater pathogen burdens. In addition, development of immune tolerance would have allowed organisms to reap the benefits of having a robust commensal microbiome, such as increased nutrient absorption and decreased colonization by pathogenic bacteria.
Though it seems that the existence of tolerance is mostly adaptive, allowing an adjustment of the immune response to a level appropriate for the given stressor, it comes with important evolutionary disadvantages. Some infectious microbes take advantage of existing mechanisms of tolerance to avoid detection and/or elimination by the host immune system. Induction of regulatory T cells, for instance, has been noted in infections with Helicobacter pylori, Listeria monocytogenes, Brugia malayi, and other worms and parasites.[8] Another important disadvantage of the existence of tolerance may be susceptibility to cancer progression. Treg cells inhibit anti-tumor NK cells.[36] The injection of Treg cells specific for a tumor antigen also can reverse experimentally-mediated tumor rejection based on that same antigen.[37] The prior existence of immune tolerance mechanisms due to selection for its fitness benefits facilitates its utilization in tumor growth.
Tradeoffs between immune tolerance and resistance
Immune tolerance contrasts with resistance. Upon exposure to a foreign antigen, either the antigen is eliminated by the standard immune response (resistance), or the immune system adapts to the pathogen, promoting immune tolerance instead.
Resistance typically protects the host at the expense of the parasite, while tolerance reduces harm to the host without having any direct negative effects on the parasite.[35] Each strategy has its unique costs and benefits for host fitness:[34]
| Costs | Benefits | |
|---|---|---|
| Elimination (resistance) |
|
|
| Tolerance |
|
|
Evolution works to optimize host fitness, so whether elimination or tolerance occurs depends on which would benefit the organism most in a given scenario. If the antigen is from a rare, dangerous invader, the costs of tolerating its presence are high and it is more beneficial to the host to eliminate it. Conversely, if experience (of the organism or its ancestors) has shown that the antigen is innocuous, then it would be more beneficial to tolerate the presence of the antigen rather than pay the costs of inflammation.
Despite having mechanisms for both immune resistance and tolerance, any one organism may be overall more skewed toward a tolerant or resistant phenotype depending on individual variation in both traits due to genetic and environmental factors.[35] In mice infected with malaria, different genetic strains of mice fall neatly along a spectrum of being more tolerant but less resistant or more resistant but less tolerant.[38] Patients with autoimmune diseases also often have a unique gene signature and certain environmental risk factors that predispose them to disease.[2] This may have implications for current efforts to identify why certain individuals may be disposed to or protected against autoimmunity, allergy, inflammatory bowel disease, and other such diseases.
References
- Medawar, Peter (December 12, 1960). "Nobel Lecture: Immunological Tolerance". The Nobel Prize. Retrieved 24 July 2020.
- Murphy, Kenneth (2012). Janeway's Immunobiology: 8th ed. Chapter 15: Garland Science. pp. 611–668. ISBN 0815342438.CS1 maint: location (link)
- Choi, J; Kim ST; Craft J (Dec 2012). "The pathogenesis of systemic lupus erythematosus-an update". Curr Opin Immunol. 24 (6): 651–657. doi:10.1016/j.coi.2012.10.004. PMC 3508331. PMID 23131610.
- Round, JL; O'Connell RM; Mazmanian SK (May 2010). "Coordination of tolerogenic immune responses by the commensal microbiota". J Autoimmun. 34 (3): J220–J225. doi:10.1016/j.jaut.2009.11.007. PMC 3155383. PMID 19963349.
- Perniola, R (2012). "Expression of the autoimmune regulator gene and its relevance to the mechanisms of central and peripheral tolerance". Clin Dev Immunol. 2012: 207403. doi:10.1155/2012/207403. PMC 3485510. PMID 23125865.
- Verbsky, JW; Chatila TA. (Dec 2013). "Immune dysregulation, polyendocrinopathy, enteropathy X-linked (IPEX) and IPEX-related disorders: an evolving web of heritable autoimmune diseases". Curr Opin Pediatr. 25 (6): 708–714. doi:10.1097/mop.0000000000000029. PMC 4047515. PMID 24240290.
- Maazi, H; Lam J; Lombardi V; Akbari O (Jun 2013). "Role of plasmacytoid dendritic cell subsets in allergic asthma". Allergy. 68 (6): 695–701. doi:10.1111/all.12166. PMC 3693732. PMID 23662841.
- Curotto de Lafaille, MA; Lafaillle JJ (2009). "Natural and Adaptive Foxp3+ Regulatory T Cells: More of the Same or a Division of Labor?". Immunity. 30 (6): 626–635. doi:10.1016/j.immuni.2009.05.002.
- Becker, JC; Andersen MH; Schrama D; Straten P (Jul 2013). "Immune-suppressive properties of the tumor microenvironment". Cancer Immunol Immunother. 62 (7): 1137–1148. doi:10.1007/s00262-013-1434-6.
- Murphy, Kenneth (2012). Janeway's Immunobiology: 8th ed. Chapter 1: Garland Sciences. pp. 13–15.CS1 maint: location (link)
- Fenner, F (1983). "The Florey lecture". Proc R Soc Lond B Biol Sci.
- Felton, LD; Kauffmann G; Prescott B; Ottinger B (Jan 1955). "Studies on the mechanism of the immunological paralysis induced in mice by pneumococcal polysaccharides". J Immunol. 74 (1): 17–26.
- Jump, RL; Levine AD (Jul 2004). "Mechanisms of Natural Tolerance in the Intestine - Implications for Inflammatory Bowel Disease". Inflamm Bowel Dis. 10 (4): 462–478. doi:10.1097/00054725-200407000-00023.
- Sprent, J; Kishimoto H (2001). "The thymus and central tolerance". Philos Trans R Soc Lond B Biol Sci. 356 (1409): 609–616. doi:10.1098/rstb.2001.0846. PMC 1088448. PMID 11375064.
- Hogquist, K; Baldwin T; Jameson S (2005). "Central tolerance: learning self-control in the thymus". Nat Rev Immunol. 5 (10): 772–782. doi:10.1038/nri1707. PMID 16200080.
- Murphy, Kenneth (2012). Janeway's Immunobiology: 8th ed. Chapter 8: Garland Sciences. pp. 275–334.CS1 maint: location (link)
- Sakaguchi, S; Miyara M; Costantino C; Hafler DA (2010). "Foxp3+ regulatory T cells in the human immune system". Nat Rev Immunol. 10: 490–500. doi:10.1038/nri2785.
- Vadasz, Z; Haj T; Kessel A; Toubi E. (Jun 2013). "B-regulatory cells in autoimmunity and immune mediated inflammation". FEBS Lett. 587 (13): 2074–2078. doi:10.1016/j.febslet.2013.05.023. PMID 23707422.
- Ganguly, D; Haak S; Sisirak V; Reizis B (2013). "The role of dendritic cells in autoimmunity". Nat Rev Immunol. 13: 566–577. doi:10.1038/nri3477. PMC 4160805. PMID 23827956.
- Maher, S; Toomey D; Condron C; Bouchier-Hayes D (Apr 2002). "Activation-induced cell death: the controversial role of Fas and Fas ligand in immune privilege and tumour counterattack". Immunol Cell Biol. 80 (2): 131–137. doi:10.1046/j.1440-1711.2002.01068.x.
- Sakaguchi, S; Wing K; Onishi Y; Prieto-Martin P; Yamaguchi T. (2009). "Regulatory T cells: how do they suppress immune responses?". Int. Immunol. 21 (10): 1105–1111. doi:10.1093/intimm/dxp095.
- Braza, F; Soulillou JP; Brouard S. (Sep 2012). "Gene expression signature in transplantation tolerance". Clin Chim Acta. 413 (17–18): 1414–1418. doi:10.1016/j.cca.2012.04.024.
- Gokmen, R; Hernandez-Fuentes MP (Aug 2013). "Biomarkers of tolerance". Curr Opin Organ Transplant. 18 (4): 416–420.
- Clark, DA; Chaouat G (2012). "Regulatory T cells and reproduction: how do they do it?". J Reprod Immunol. 96 (1–2): 1–7. doi:10.1016/j.jri.2012.07.007.
- Christiansen, OB (2013). "Reproductive immunology". Mol Immunol. 55 (1): 8–16. doi:10.1016/j.molimm.2012.08.025. PMC 1383872.
- Murphy, K (2012). Janeway's Immunobiology: 8th ed. Chapter 12: Garland Sciences. pp. 465–502.CS1 maint: location (link)
- Wiener, HL (2000). "Oral tolerance, an active immunologic process mediated by multiple mechanisms". J Clin Invest. 106 (8): 935–937. doi:10.1172/jci11348. PMC 314352. PMID 11032852.
- Soyer, OU; Akdis M; Ring J; Behrendt H; Crameri R; Lauener R; Akdis CA (2012). "Mechanisms of peripheral tolerance to allergens". Allergy. 68 (2): 161–170. doi:10.1111/all.12085.
- Petalas, K; Durham SR (2013). "Allergen immunotherapy for allergic rhinitis". Rhinology. 51 (2): 99–110.
- Cernadas, JR (Feb 2013). "Desensitization to antibiotics in children". Pediatr Allergy Immunol. 24 (1): 3–9. doi:10.1111/pai.12001.
- Aktipis, CA; Boddy AM; Getnby RA; Brown JS; Maley CC (2013). "Life history trade-offs in cancer evolution". Nat Rev Cancer. 13 (12): 883–892. doi:10.1038/nrc3606. PMC 4010142. PMID 24213474.
- Ramsay, AG (2013). ". Immune checkpoint blockade immunotherapy to activate anti-tumour T-cell immunity". Br J Haematol. 162 (3): 313–325. doi:10.1111/bjh.12380.
- Lindau, D; Gielen P; Kroesen M; Wesseling P; Adema GJ (2013). "The immunosuppressive tumor network: myeloid-derived suppressor cells, regulatory T cells, and natural killer T cells". Immunology. 138 (2): 105–115. doi:10.1111/imm.12036. PMC 3575763. PMID 23216602.
- Medzhitov, R; Schneider DS; Soares MP (Feb 24, 2012). "Disease Tolerance as a Defense Strategy". Science. 335 (6071): 936–941. doi:10.1126/science.1214935. PMC 3564547. PMID 22363001.
- Raberg, L; Graham AL; Read AF (Jan 12, 2009). "Decomposing health: tolerance and resistance to parasites in animals". Philos Trans R Soc Lond B Biol Sci. 364 (1513): 37–49. doi:10.1098/rstb.2008.0184. PMC 2666700. PMID 18926971.
- Ghiringhelli, F; Ménard C; Martin F; Zitvogel L (Dec 2006). "The role of regulatory T cells in the control of natural killer cells: relevance during tumor progression". Immunol Rev. 214: 229–238. doi:10.1111/j.1600-065x.2006.00445.x.
- Kretschmer, K; Apostolou I; Jaeckel E; Khazaie K; von Boehmer H (Aug 2006). "Making regulatory T cells with defined antigen specificity: role in autoimmunity and cancer". Immunol Rev. 212: 163–169. doi:10.1111/j.0105-2896.2006.00411.x.
- Raberg, L; Sim D; Read AF (2007). "Disentangling Genetic Variation for Resistance and Tolerance to Infectious Diseases in Animals". Science. 318: 812–814. doi:10.1126/science.1148526. PMID 17975068.
External links
- Immune Tolerance Network
- International Conference on Immune Tolerance
- Immune+tolerance at the US National Library of Medicine Medical Subject Headings (MeSH)