Seipin
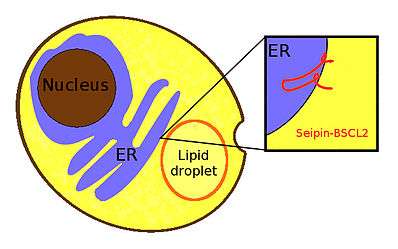
Seipin is a homo-oligomeric integral membrane protein in the endoplasmic reticulum (ER) that concentrates at junctions with cytoplasmic lipid droplets (LDs). Alternatively, seipin can be referred to as Bernardinelli-Seip congenital lipodystrophy type 2 protein (BSCL2), and it is encoded by the corresponding gene of the same name, i.e. BSCL2. At protein level, seipin is expressed in cortical neurons in the frontal lobes, as well as motor neurons in the spinal cord. It is highly expressed in areas like the brain, testis and adipose tissue.[1] Seipin's function is still unclear but it has been localized close to lipid droplets, and cells knocked out in seipin which have anomalous droplets.[2] Hence, recent evidence suggests that seipin plays a crucial role in lipid droplet biogenesis.[3]
| Putative adipose-regulatory protein | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | Seipin | ||||||||
| Pfam | PF06775 | ||||||||
| InterPro | IPR009617 | ||||||||
| OPM superfamily | 526 | ||||||||
| OPM protein | 6ds5 | ||||||||
| |||||||||
Function
Though it was initially dubbed "mysterious protein",[4] recent empirical studies are gradually starting to unveil some of seipin's most compelling physiological functions.[5][6][7][8][9] Among these, the following have been identified: central regulation of energy homeostasis, lipid catabolism (essential for adipocyte differentiation), lipid storage and lipid droplet maintenance, as well as prevention of ectopic lipid droplet formation in non-adipose tissues.[10] Additionally, mutations of BSCL2 have been recently linked to the Silver-Syndrome[11] and Celia's Encephalopathy.[12]
Structure
Primary structure
The seipin gene BSCL2 was originally identified in mammals and the fruit fly, and later extended to fungi and plants.[13] The human seipin gene is located on chromosome 11q13, with protein coding on the Crick strand.[14]
There are three validated coding transcripts in GenBank. The primary transcript originally described, contained 11 exons with protein coding beginning on exon 2 and ending in exon 11 (transcript variant 2), resulting in a 398 amino acid protein with two strongly predicted transmembrane domains (TMDs), coded in exons 2 and 7 (isoform 2).
However, a longer transcript (variant 1) is generated with an alternative first exon containing a translational start site that results in an additional 64 amino acids at the N-terminal extension, 462 amino acids in total (isoform 1).
A third coding transcript (variant 3) splices out exon 7 and produces a shortened and altered carboxy terminus in exon 10, generating a protein of 287 amino acids (isoform 3).[3]
Secondary structure
The secondary structure of seipin includes a conserved central core domain, and diverse cytosolic N- and C-termini.[15]
Tertiary structure
The protein has a short cytoplasmic region, a transmembrane alpha-helix, a water-soluble beta-sandwich domain located in endoplasmic reticulum, and second TM helix.
Pathophysiology
There are three different variations of seipin's amino acid sequence:[16]
- a N → S change in position 88, does not affect protein subcellular location.
- a S → L change in position 90, does not affect the function in lipid storage.
- a A → P change in position 212 that increases localization to nuclear envelope.
All seipin mutations occur within its loop domain. Between some of these, four large deletions can be found which indicate that at least exons 4 and 5 are required for seipin function in humans. In addition, other six mutations have been identified in the loop domain. The majority of these cluster at the single asparagine-linked glycosylation site (NVS) in seipin.[3] The two mutations that cause neuronal seipinopathy, N88S and S90L, are located directly within this site.[17] Apart from suspending the glycosylation process, these mutations engender an aggregation of seipin and, consequently, the initiation of the ER stress response. The seipin protein can also have a modification residue, that can transform the 289’ and 372’ serine into a phosphoserine, an ester of serine and phosphoric acid.
Overexpression of mutated seipin proteins N88S or S90L can also activate autophagy, and substantially altering the sub-cellular distribution of the autophagosome marker GFP-LC3, which leads to a number of large vacuoles appearing in the cytoplasm. The sub-cellular location of GFP-LC3 and mutated seipin proteins highly overlap. Moreover, these seipin proteins can diffuse small lipid droplets to fuse into larger lipid.
Seipin mutations have been associated with congenital generalized lipodystrophy (see below), and mutations in an N-glycosylation motif links seipin to two other disorders, i.e. Silver syndrome and autosomal-dominant distal hereditary motor neuropathy type V.[18]
Disease associations
Congenital generalized lipodystrophy
CGL (congenital generalized lipodystrophy) is a heterogeneous genetic disorder characterized by almost complete loss of adipose tissue (both metabolic and mechanical adipose depots) and an increase of ectopic fat storage in liver and muscle. Of the four CGL types, BSCL2 (Berardinelli-Seip Congenital lipodystrophy type 2), resulting from mutations in the BSCL2/seipin gene, exhibits the most severe lipodystrophic phenotype.[19]
Furthermore, these patients could suffer dyslipidemia, hepatic steatosis, insulin resistance and hypertrophic cardiomyopathy due to a cell-autonomous defect in cardiomyocytes.[3]
Neurological seipinopathies
For many years mutations of the seipin gene were associated with a loss of function, such as in CGL (see above). However, recent studies show that mutations such as N88S and S90L seem to have a gain-of-toxic-function which may result in autosomal dominant motor neuron diseases and distal hereditary motor neuropathy.
Owing to the wide clinical spectrum of these mutations, it has been proposed to collectively refer to seipin-related motor neuron diseases as seipinopathies.[20]
Symptoms can vary and include: developmental regression of motor and cognitive skills in the first years of life leading to death (encephalopathy), muscle weakness and spasticity in lower limbs (spastic paraplegia type XVII), weakness of distal muscles of upper limbs (distal hereditary motor neuropathy type V) as well as wasting of the hand muscles (in both cases). Complex forms of seipinopathies may include deafness, dementia or mental retardation.[3]
Male infertility
Testicular tissue-derived seipin is essential for male fertility by modulating testicular phospholipid homeostasis. The lack of seipin in germ cells results in complete male infertility and teratozoospermia. Spermatids devoid of seipin in germ cells are morphologically abnormal with large ectopic lipid droplets and aggregate in dysfunctional clusters. Elevated levels of phosphatidic acid accompanied with an altered ratio of polyunsaturated to monounsaturated and saturated fatty acids show impaired phospholipid homeostasis during spermiogenesis[21] .
See also
- UniProtKB - Q96G97 (BSCL2_HUMAN)
- Common Therapies in Lipodystrophy Treatment
- Silver–Russell syndrome
- Endoplasmic Reticulum Stress in Beta Cells
- Muscle Weakness
- Distal hereditary motor neuronopathies
- Upper motor neuron lesion
References
- "MobiDB". mobidb.bio.unipd.it. Retrieved 2015-10-28.
- Fan HD, Chen SP, Sun YX, Xu SH, Wu LJ (April 2015). "Seipin mutation at glycosylation sites activates autophagy in transfected cells via abnormal large lipid droplets generation". Acta Pharmacologica Sinica. 36 (4): 497–506. doi:10.1038/aps.2014.164. PMC 4387305. PMID 25832430.
- Cartwright BR, Binns DD, Hilton CL, Han S, Gao Q, Goodman JM (February 2015). "Seipin performs dissectible functions in promoting lipid droplet biogenesis and regulating droplet morphology". Molecular Biology of the Cell. 26 (4): 726–39. doi:10.1091/mbc.E14-08-1303. PMC 4325842. PMID 25540432.
- Agarwal AK, Garg A (September 2004). "Seipin: a mysterious protein". Trends in Molecular Medicine. 10 (9): 440–4. doi:10.1016/j.molmed.2004.07.009. PMID 15350896.
- Fan HD, Chen SP, Sun YX, Xu SH, Wu LJ (April 2015). "Seipin mutation at glycosylation sites activates autophagy in transfected cells via abnormal large lipid droplets generation". Acta Pharmacologica Sinica. 36 (4): 497–506. doi:10.1038/aps.2014.164. PMC 4387305. PMID 25832430.
- Talukder MM, Sim MF, O'Rahilly S, Edwardson JM, Rochford JJ (March 2015). "Seipin oligomers can interact directly with AGPAT2 and lipin 1, physically scaffolding critical regulators of adipogenesis". Molecular Metabolism. 4 (3): 199–209. doi:10.1016/j.molmet.2014.12.013. PMC 4338318. PMID 25737955.
- Patni N, Garg A (September 2015). "Congenital generalized lipodystrophies--new insights into metabolic dysfunction". Nature Reviews. Endocrinology. 11 (9): 522–34. doi:10.1038/nrendo.2015.123. PMID 26239609.
- Cai Y, Goodman JM, Pyc M, Mullen RT, Dyer JM, Chapman KD (September 2015). "Arabidopsis SEIPIN Proteins Modulate Triacylglycerol Accumulation and Influence Lipid Droplet Proliferation". The Plant Cell. 27 (9): 2616–36. doi:10.1105/tpc.15.00588. PMC 4815042. PMID 26362606.
- Ebihara C, Ebihara K, Aizawa-Abe M, Mashimo T, Tomita T, Zhao M, et al. (August 2015). "Seipin is necessary for normal brain development and spermatogenesis in addition to adipogenesis". Human Molecular Genetics. 24 (15): 4238–49. doi:10.1093/hmg/ddv156. PMID 25934999.
- Boutet E, El Mourabit H, Prot M, Nemani M, Khallouf E, Colard O, et al. (June 2009). "Seipin deficiency alters fatty acid Delta9 desaturation and lipid droplet formation in Berardinelli-Seip congenital lipodystrophy". Biochimie. 91 (6): 796–803. doi:10.1016/j.biochi.2009.01.011. PMID 19278620.
- Monteiro A, Real R, Nadais G, Silveira F, Leão M (March 2015). "BSCL2 N88S mutation in a Portuguese patient with the Silver syndrome". Muscle & Nerve. 51 (3): 456–8. doi:10.1002/mus.24455. PMID 25219579.
- Ruiz-Riquelme A, Sánchez-Iglesias S, Rábano A, Guillén-Navarro E, Domingo-Jiménez R, Ramos A, et al. (November 2015). "Larger aggregates of mutant seipin in Celia's Encephalopathy, a new protein misfolding neurodegenerative disease". Neurobiology of Disease. 83: 44–53. doi:10.1016/j.nbd.2015.08.006. PMID 26282322.
- Fei W, Shui G, Gaeta B, Du X, Kuerschner L, Li P, Brown AJ, Wenk MR, Parton RG, Yang H (February 2008). "Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast". The Journal of Cell Biology. 180 (3): 473–82. doi:10.1083/jcb.200711136. PMC 2234226. PMID 18250201.
- Magré J, Delépine M, Khallouf E, Gedde-Dahl T, Van Maldergem L, Sobel E, et al. (August 2001). "Identification of the gene altered in Berardinelli-Seip congenital lipodystrophy on chromosome 11q13". Nature Genetics. 28 (4): 365–70. doi:10.1038/ng585. PMID 11479539.
- Yang W, Thein S, Guo X, Xu F, Venkatesh B, Sugii S, Radda GK, Han W (May 2013). "Seipin differentially regulates lipogenesis and adipogenesis through a conserved core sequence and an evolutionarily acquired C-terminus". The Biochemical Journal. 452 (1): 37–44. doi:10.1042/BJ20121870. PMID 23458123.
- Universal protein resource accession number Q96G97 for "BSCL2 - Seipin - Homo sapiens (Human) - BSCL2 gene & protein" at UniProt.
- Windpassinger C, Auer-Grumbach M, Irobi J, Patel H, Petek E, Hörl G, et al. (March 2004). "Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and Silver syndrome". Nature Genetics. 36 (3): 271–6. doi:10.1038/ng1313. PMID 14981520.
- Lundin C, Nordström R, Wagner K, Windpassinger C, Andersson H, von Heijne G, et al. (April 2006). "Membrane topology of the human seipin protein". FEBS Letters. 580 (9): 2281–4. doi:10.1016/j.febslet.2006.03.040. PMID 16574104.
- Wee K, Yang W, Sugii S, Han W (October 2014). "Towards a mechanistic understanding of lipodystrophy and seipin functions". Bioscience Reports. 34 (5): 583–591. doi:10.1042/BSR20140114. PMC 4182903. PMID 25195639.
- Ito D, Suzuki N (January 2009). "Seipinopathy: a novel endoplasmic reticulum stress-associated disease". Brain. 132 (Pt 1): 8–15. doi:10.1093/brain/awn216. PMID 18790819.
- Jiang M, Gao M, Wu C, He H, Guo X, Zhou Z, Yang H, Xiao X, Liu G, Sha J (May 2014). "Lack of testicular seipin causes teratozoospermia syndrome in men". Proceedings of the National Academy of Sciences of the United States of America. 111 (19): 7054–9. doi:10.1073/pnas.1324025111. PMC 4024893. PMID 24778225.