Kinin–kallikrein system
The kinin–kallikrein system or simply kinin system is a poorly understood hormonal system with limited available research.[1] It consists of blood proteins that play a role in inflammation,[2] blood pressure control, coagulation and pain. Its important mediators bradykinin and kallidin are vasodilators and act on many cell types.
History
The system was discovered in 1909 when researchers discovered that injection with urine (high in kinins) led to hypotension (low blood pressure).[3] The researchers Emil Karl Frey, Heinrich Kraut and Eugen Werle discovered high-molecular weight kininogen in urine around 1930.[4]
Etymology
kinin [Gk] kīn(eîn) to move, set in motion. kallikrein [Gk ] kalli~ sweet and krein = kreos, flesh, named for the pancreatic extracts where it was first discovered
Members
The system consists of a number of large proteins, some small polypeptides and a group of enzymes that activate and deactivate the compounds.
Proteins
High-molecular weight kininogen (HMWK) and low-molecular weight kininogen (LMWK) are precursors of the polypeptides. They have no activity of themselves.
- HMWK is produced by the liver together with prekallikrein (see below). It acts mainly as a cofactor on coagulation and inflammation, and has no intrinsic catalytic activity.
- LMWK is produced locally by numerous tissues, and secreted together with tissue kallikrein.
Polypeptides
- Bradykinin (BK), which acts on the B2 receptor and slightly on B1, is produced when kallikrein releases it from HMWK. It is a nonapeptide (9 amino acids) with the amino acid sequence Arg–Pro–Pro–Gly–Phe–Ser–Pro–Phe–Arg.
- Kallidin (KD) is released from LMWK by tissue kallikrein. It is a decapeptide. KD has the same amino acid sequence as Bradykinin with the addition of a Lysine at the N-terminus, thus is sometimes referred to as Lys-Bradykinin.
HMWK and LMWK are formed by alternative splicing of the same gene.[5]
Enzymes
- Kallikreins (tissue and plasma kallikrein) are serine proteases that liberate kinins[6] (BK and KD) from the kininogens, which are plasma proteins that are converted into vasoactive peptides.[7] Prekallikrein is the precursor of plasma kallikrein. It can only activate kinins after being activated itself by factor XIIa or other stimuli.
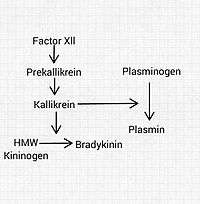
- Carboxypeptidases are present in two forms: N circulates and M is membrane-bound. They remove arginine residues at the carboxy-terminus of BK and KD.
- Angiotensin converting enzyme (ACE), also termed kininase II, inactivates a number of peptide mediators, including bradykinin. It is better known for activating angiotensin.
- Neutral endopeptidase also deactivates kinins and other mediators.
Pharmacology
Inhibition of ACE with ACE inhibitors leads to decreased conversion of angiotensin I to angiotensin II (a vasoconstrictor) but also to an increase in bradykinin due to decreased degradation. This explains why some patients taking ACE inhibitors develop a dry cough, and some react with angioedema, a dangerous swelling of the head and neck region.
There are hypotheses that many of the ACE-inhibitors' beneficial effects are due to their influence on the kinin-kallikrein system. This includes their effects in arterial hypertension, in ventricular remodeling (after myocardial infarction) and possibly diabetic nephropathy.
Role in disease
Defects of the kinin-kallikrein system in diseases are not generally recognized. The system is the subject of much research due to its relationship to the inflammation and blood pressure systems. It is known that kinins are inflammatory mediators that cause dilation of blood vessels and increased vascular permeability. Kinins are small peptides produced from kininogen by kallikrein and are broken down by kininases. They act on phospholipase and increase arachidonic acid release and thus prostaglandin (PGE2) production.
C1-INH Involvement
C1-inhibitor is a serine protease inhibitor (serpin) protein. C1-INH is the most important physiological inhibitor of plasma kallikrein, fXIa and fXIIa. C1-INH also inhibits proteinases of the fibrinolytic, clotting, and kinin pathways. Deficiency of C1-INH permits plasma kallikrein activation, which leads to the production of the vasoactive peptide bradykinin.
References
- Seth (1 January 2008). Textbook Of Pharmacology. Elsevier India. pp. 603–. ISBN 978-81-312-1158-8. Retrieved 25 November 2010.
- Duchene (2011). "Kallikrein-kinin kystem in inflammatory diseases". Kinins. De Gruyter. pp. 261–272. ISBN 978-3-11-025235-4.
- Abelous JE, Bardier E (1909). "Les substances hypotensives de l'urine humaine normale". CR Soc Biol (in French). 66: 511–20.
- Kraut H, Frey EK, Werle E (1930). "Der Nachweis eines Kreislaufhormon in der Pankreasdrüse". Hoppe-Seyler's Z Physiol Chem. 189 (3–4): 97–106. doi:10.1515/bchm2.1930.189.3-4.97.
- Goodman & Gilman's Pharmacology; Chapter 24. Histamine, Bradykinin, and Their Antagonists
- Stefan Offermanns; Walter Rosenthal (2008). Encyclopedia of Molecular Pharmacology. Springer. pp. 673–. ISBN 978-3-540-38916-3. Retrieved 11 December 2010.
- Kumar, V., Abbas, A., Fausto, N. (Editors) Robbins and Cotran pathologic basis of disease. 7th ed. Philadelphia: Elsevier 2005;Page 65.
External links
- Kallikrein-Kinin+System at the US National Library of Medicine Medical Subject Headings (MeSH)