I-cell disease
Inclusion-cell (I-cell) disease, also referred to as mucolipidosis II (ML II),[1][2] is part of the lysosomal storage disease family and results from a defective phosphotransferase (an enzyme of the Golgi apparatus). This enzyme transfers phosphate to mannose residues on specific proteins. Mannose-6-phosphate serves as a marker for proteins to be targeted to lysosomes within the cell. Without this marker, proteins are instead secreted outside the cell, which is the default pathway for proteins moving through the Golgi apparatus. Lysosomes cannot function without these proteins, which function as catabolic enzymes for the normal breakdown of substances (e.g. oligosaccharides, lipids, and glycosaminoglycans)[3] in various tissues throughout the body (i.e. fibroblasts). As a result, a buildup of these substances occurs within lysosomes because they cannot be degraded, resulting in the characteristic I-cells, or "inclusion cells" seen microscopically. In addition, the defective lysosomal enzymes normally found only within lysosomes are instead found in high concentrations in the blood, but they remain inactive at blood pH (around 7.4) because they require the low lysosomal pH 5 to function.
| I-cell disease | |
|---|---|
| Other names | Mucolipidosis II (ML II) |
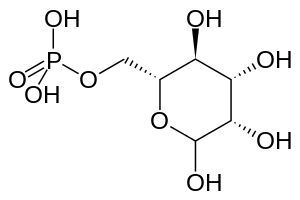 | |
| Mannose-6-phosphate (M6P). I-cell disease involves a failure to add M6P to proteins. | |
| Specialty | Medical genetics |
Signs and symptoms
Mucolipidosis II (ML II) is a particularly severe form of ML that has a significant resemblance to another mucopolysaccharidosis called Hurler syndrome. Generally only laboratory testing can distinguish the two as the presentation is so similar, with high plasma concentrations of lysosomal enzymes, often fatal in childhood.[4] Typically, by the age of 6 months, failure to thrive and developmental delays are obvious signs of this disorder. Some physical signs, such as abnormal skeletal development, coarse facial features, and restricted joint movement, may be present at birth. Children with ML II usually have enlargement of certain organs, such as the liver (hepatomegaly) or spleen (splenomegaly), and sometimes even the heart valves. Affected children often have stiff claw-shaped hands and fail to grow and develop in the first months of life. Delays in the development of their motor skills are usually more pronounced than delays in their cognitive (mental processing) skills. Children with ML II eventually develop a clouding on the cornea of their eyes and, because of their lack of growth, develop short-trunk dwarfism (underdeveloped trunk). These young patients are often plagued by recurrent respiratory tract infections, including pneumonia, otitis media (middle ear infections), bronchitis and carpal tunnel syndrome. Children with ML II generally die before their seventh year of life, often as a result of congestive heart failure or recurrent respiratory tract infections.
Pathophysiology
I-cell disease is an autosomal recessive disorder caused by a deficiency of GlcNAc phosphotransferase, which phosphorylates mannose residues to mannose-6-phosphate on N-linked glycoproteins in the Golgi apparatus within cells. Without mannose-6-phosphate to target them to the lysosomes, the enzymes are erroneously transported from the Golgi to the extracellular space. Consequently lysosomes lack the requisite hydrolytic enzymes needed for catabolism of cellular debris, so this debris accumulates within them and forms the characteristic intracellular inclusions (hence the name of the disorder).[5] Hydrolases secreted into the blood stream cause little problem as they are inactivate at the near neutral pH of blood (7.4).
It can be associated with N-acetylglucosamine-1-phosphate transferase (GNPTA).[6] In a case report, I-cell disease was complicated by severe dilative cardiomyopathy (DCM).[7]
Though rare, a deficiency of phosphodiesterase which would cleave GlcNAc from the mannose-6-phosphate tag will also cause I-cell disease.[5] The presence of lipids, glycosaminoglycans (GAG's) and carbohydrates in the blood provide for the distinguishing characteristic to separate I-cell from Hurler Syndrome. In Hurler's, only glycosaminoglycans would be present.
Diagnosis
Diagnostic measures can include the following.
Before birth:
- Abnormally low concentrations of UDP-N-acetylglucosamine-1-phosphotransferase enzyme activity in amniotic fluid cells or chorionic villi [8]
In infants:
- Elevated plasma lysosomal enzyme concentrations [8]
- Decreased concentration of lysosomal enzymes in cultured fibroblasts [8]
- Presence of inclusion bodies in peripheral blood lymphocytes [8]
- Low concentrations of UDP-N-acetylglucosamine-1-phosphotransferase enzyme activity as measured in white blood cells [8]
Treatment
There is no cure for I-Cell disease/Mucolipidosis II disease; treatment is limited to controlling or reducing symptoms. Nutritional supplements, particularly iron and vitamin B12, are often recommended. Physical therapy to improve motor delays and speech therapy to improve language acquisition are treatment options. Surgery can remove the thin layer of corneal clouding to temporarily improve the complication. It is possible that bone marrow transplant may be helpful in delaying or correcting the neurological deterioration that occurs with I-Cell disease.[9]. The Yash Gandhi Foundation is a US non-profit organization which funds research for I-Cell disease [10]
References
- "mucolipidosis II" at Dorland's Medical Dictionary
- Plante M, Claveau S, Lepage P, et al. (March 2008). "Mucolipidosis II: a single causal mutation in the N-acetylglucosamine-1-phosphotransferase gene (GNPTAB) in a French Canadian founder population". Clin. Genet. 73 (3): 236–44. doi:10.1111/j.1399-0004.2007.00954.x. PMID 18190596.
- Bamshad, Lynn B. Jorde, John C. Carey, Michael J. (2010). Medical genetics (4th ed.). Philadelphia: Mosby/Elsevier. ISBN 9780323053730.
- Le, Tao (2014). First Aid for the USMLE 2014. New York: McGraw Hill Education. p. 77. ISBN 9780071831420.
- Champe, Pamela (2004). Lippincott's Illustrated Reviews: Biochemistry. Richard A Harvey, Denise R Ferrier (3rd ed.). Philadelphia, Pa.: Lippincott-Raven. p. 167. ISBN 978-0-7817-2265-0.
- Tiede S, Storch S, Lübke T, et al. (2005). "Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase". Nat. Med. 11 (10): 1109–12. doi:10.1038/nm1305. PMID 16200072.
- Sahha.gov.mt - 2006 Dec;29_1
- "I Cell Disease - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders). Retrieved 2017-11-02.
- "Archived copy". Archived from the original on 2010-06-20. Retrieved 2009-12-01.CS1 maint: archived copy as title (link)
- "Yash Gandhi Foundation".
External links
| Classification | |
|---|---|
| External resources |
- mucolipidoses at NINDS — article derived from detail sheet available here
- I cell disease at NIH's Office of Rare Diseases
- GeneReview/NIH/UW entry on Mucolipidosis II