Hypertryptophanemia
Hypertryptophanemia, is a rare autosomal recessive[2] metabolic disorder that results in a massive buildup of the amino acid tryptophan in the blood, with associated symptoms and tryptophanuria (-uria denotes "in the urine").[3][4]
| Hypertryptophanemia | |
|---|---|
| Other names | Familial hypertryptophanemia[1] |
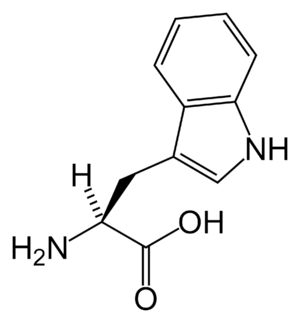 | |
| Tryptophan | |
| Specialty | Endocrinology |
Elevated levels of tryptophan are also seen in Hartnup disease,[5] a disorder of amino acid transport.[6] However, the increase of tryptophan in that disorder is negligible when compared to that of hypertryptophanemia.[1][5]
Symptoms
A number of abnormalities and symptoms have been observed with hypertryptophanemia.
Musculoskeletal effects include: joint contractures of the elbows and interphalangeal joints of the fingers and thumbs (specifically the distal phalanges), pes planus (fallen arches), an ulnar drift affecting the fingers of both hands (an unusual, yet correctible feature where the fingers slant toward the ulnar side of the forearm), joint pain and laxity, and adduction of the thumbs (where the thumb appears drawn into the palm, related to contracture of the adductor pollicis).[1][2]
Behavioral, developmental and other anomalies often include: hypersexuality, perceptual hypersensitivity, emotional lability (mood swings),[3] hyperaggressive behavior;[2] hypertelorism (widely-set eyes), optical strabismus (misalignment) and myopia.[1][2]
Metabolically, hypertryptophanemia results in tryptophanuria and exhibits significantly elevated serum levels of tryptophan, exceeding 650% of maximum (normal range: 25-73 micromole/l) in some instances.[2][3]
A product of the bacterial biosynthesis of tryptophan is indole.[7][8] The excess of tryptophan in hypertryptophanemia also results in substantial excretion of indoleic acids. These findings suggest a possible congenital defect in the metabolic pathway where tryptophan is converted to kynurenine.[3]
Genetics
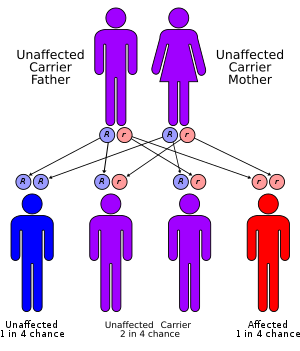
Hypertryptophanemia is believed to be inherited in an autosomal recessive manner.[2] This means a defective gene responsible for the disorder is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
Pathophysiology
At present, no specific enzyme deficiency nor genetic mutation has been implicated as the cause of hypertryptophanemia.[1][2] Several known factors regarding tryptophan metabolism and kynurenines, however, may explain the presence of behavioral abnormalities seen with the disorder.
Tryptophan is an essential amino acid, and is required for protein synthesis.[9] Aside from this crucial role, the remainder of tryptophan is primarily metabolized along the kynurenine pathway in most tissues, including those of the brain and central nervous system.[10][11][12][13]
As the main defect behind hypertryptophanemia is suspected to alter and disrupt the metabolic pathway from tryptophan to kynurenine,[2] a possible correlation between hypertryptophanemia and the known effects of kynurenines on neuronal function, physiology and behavior may be of interest.[14][15]
One of these kynurenines, aptly named kynurenic acid, serves as a neuroprotectant through its function as an antagonist at both nicotinic and glutamate receptors (responsive to the neurotransmitters nicotine and glutamate, respectively).[11][12] This action is in opposition to the agonist quinolinic acid, another kynurenine, noted for its potential as a neurotoxin.[10][13] Quinolinic acid activity has been associated with neurodegenerative disorders such as Huntington's disease, the neuroprective abilities of kynurenic acid forming a counterbalance against this process, and the related excitotoxicity and similar damaging effects on neurons.[13][14]
Indoleic acid excretion is another indicator of hypertryptophanemia.[2][3] Indirectly related to kynurenine metabolism, indole modifies neural function and human behavior by interacting with voltage-dependent sodium channels (integral membrane proteins that form ion channels, allowing vital synaptic action potentials).[15]
Diagnosis
Management
See also
References
- Online Mendelian Inheritance in Man (OMIM): 600627
- Martin JR, Mellor CS, Fraser FC (April 1995). "Familial hypertryptophanemia in two siblings". Clin. Genet. 47 (4): 180–183. doi:10.1111/j.1399-0004.1995.tb03956.x. PMID 7628119.
- Snedden W, Mellor CS, Martin JR (July 1983). "Familial hypertryptophanemia, tryptophanuria and indoleketonuria". Clinica Chimica Acta. 131 (3): 247–256. doi:10.1016/0009-8981(83)90094-3. ISSN 0009-8981. PMID 6883719.
- Snedden W, Mellor CS, Martin JR (November 1982). "Hypertryptophanemia and indoleketonuria in two mentally subnormal siblings" (Free full text). The New England Journal of Medicine. 307 (22): 1405. doi:10.1056/NEJM198211253072219. ISSN 0028-4793. PMID 7133092.
- Online Mendelian Inheritance in Man (OMIM): 234500
- Seow HF, Bröer S, Bröer A, Bailey CG, Potter SJ, Cavanaugh JA, Rasko JE (September 2004). "Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19". Nature Genetics. 36 (9): 1003–1007. doi:10.1038/ng1406. PMID 15286788.
- Dunn MF, Niks D, Ngo H, Barends TR, Schlichting I (June 2008). "Tryptophan synthase: the workings of a channeling nanomachine". Trends in Biochemical Sciences. 33 (6): 254–264. doi:10.1016/j.tibs.2008.04.008. PMID 18486479.
- Houben KF, Dunn MF (March 1990). "Allosteric effects acting over a distance of 20-25 A in the Escherichia coli tryptophan synthase bienzyme complex increase ligand affinity and cause redistribution of covalent intermediates". Biochemistry. 29 (9): 2421–2429. doi:10.1021/bi00461a028. ISSN 0006-2960. PMID 2186812.
- Reeds PJ (1 July 2000). "Dispensable and indispensable amino acids for humans" (Free full text). The Journal of Nutrition. 130 (7): 1835S–1840S. doi:10.1093/jn/130.7.1835S. ISSN 0022-3166. PMID 10867060.
- Stone TW (January 2001). "Endogenous neurotoxins from tryptophan". Toxicon. 39 (1): 61–73. doi:10.1016/S0041-0101(00)00156-2. ISSN 0041-0101. PMID 10936623.
- Stone TW, Mackay GM, Forrest CM, Clark CJ, Darlington LG (July 2003). "Tryptophan metabolites and brain disorders". Clinical Chemistry and Laboratory Medicine. 41 (7): 852–859. doi:10.1515/CCLM.2003.129. PMID 12940508.
- Stone TW, Forrest CM, Mackay GM, Stoy N, Darlington LG (December 2007). "Tryptophan, adenosine, neurodegeneration and neuroprotection". Metabolic Brain Disease. 22 (3–4): 337–352. doi:10.1007/s11011-007-9064-3. PMID 17712616.
- Stone TW (April 2001). "Kynurenic acid antagonists and kynurenine pathway inhibitors". Expert Opin Investig Drugs. 10 (4): 633–645. doi:10.1517/13543784.10.4.633. PMID 11281814.
- Ruddick JP, Evans AK, Nutt DJ, Lightman SL, Rook GA, Lowry CA (August 2006). "Tryptophan metabolism in the central nervous system: medical implications". Expert Reviews in Molecular Medicine. 8 (20): 1–27. doi:10.1017/S1462399406000068. PMID 16942634.
- Moroni F (June 1999). "Tryptophan metabolism and brain function: focus on kynurenine and other indole metabolites". European Journal of Pharmacology. 375 (1–3): 87–100. doi:10.1016/S0014-2999(99)00196-X. ISSN 0014-2999. PMID 10443567.
External links
| Classification | |
|---|---|
| External resources |