Genome evolution
Genome evolution is the process by which a genome changes in structure (sequence) or size over time. The study of genome evolution involves multiple fields such as structural analysis of the genome, the study of genomic parasites, gene and ancient genome duplications, polyploidy, and comparative genomics. Genome evolution is a constantly changing and evolving field due to the steadily growing number of sequenced genomes, both prokaryotic and eukaryotic, available to the scientific community and the public at large.

History
Since the first sequenced genomes became available in the late 1970s,[1] scientists have been using comparative genomics to study the differences and similarities between various genomes. Genome sequencing has progressed over time to include more and more complex genomes including the eventual sequencing of the entire human genome in 2001.[2] By comparing genomes of both close relatives and distant ancestors the stark differences and similarities between species began to emerge as well as the mechanisms by which genomes are able to evolve over time.
Prokaryotic and eukaryotic genomes
Prokaryotes
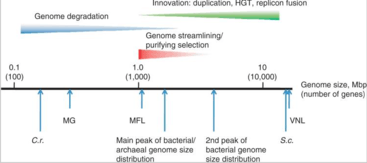
Prokaryotic genomes have two main mechanisms of evolution: mutation and horizontal gene transfer.[3] A third mechanism, sexual reproduction, prominent in eukaryotes, is not found in bacteria although prokaryotes can acquire novel genetic material through the process of bacterial conjugation in which both plasmids and whole chromosomes can be passed between organisms. An often cited example of this process is the transfer of antibiotic resistance utilizing plasmid DNA.[4] Another mechanism of genome evolution is provided by transduction whereby bacteriophages introduce new DNA into a bacterial genome.
Genome evolution in bacteria is well understood because of the thousands of completely sequenced bacterial genomes available. Genetic changes may lead to both increases or decreases of genomic complexity due to adaptive genome streamlining and purifying selection.[5] In general, free-living bacteria have evolved larger genomes with more genes so they can adapt more easily to changing environmental conditions. By contrast, most parasitic bacteria have reduced genomes as their hosts supply many if not most nutrients, so that their genome does not need to encode for enzymes that produce these nutrients themselves.[6]
| Characteristic | E.coli genome | Human genome |
|---|---|---|
| Genome Size (base pairs) | 4.6 Mb | 3.2 Gb |
| Genome Structure | Circular | Linear |
| Number of chromosomes | 1 | 46 |
| Presence of Plasmids | Yes | No |
| Presence of Histones | No | Yes |
| DNA segregated in the nucleus | No | Yes |
| Number of genes | 4,288 | 20,000 |
| Presence of Introns | No* | Yes |
| Average Gene Size | 700 bp | 27,000 bp |
Eukaryotes
Eukaryotic genomes are generally larger than that of the prokaryotes. While the E. coli genome is roughly 4.6Mb in length,[8] in comparison the Human genome is much larger with a size of approximately 3.2Gb.[9] The eukaryotic genome is linear and can be composed of multiple chromosomes, packaged in the nucleus of the cell. The non-coding portions of the gene, known as introns, which are largely not present in prokaryotes, are removed by RNA splicing before translation of the protein can occur. Eukaryotic genomes evolve over time through many mechanisms including sexual reproduction which introduces much greater genetic diversity to the offspring than the prokaryotic process of replication in which the offspring are theoretically genetic clones of the parental cell.
Genome size
Genome size is usually measured in base pairs (or bases in single-stranded DNA or RNA). The C-value is another measure of genome size. Research on prokaryotic genomes shows that there is a significant positive correlation between the C-value of prokaryotes and the amount of genes that compose the genome.[10] This indicates that gene number is the main factor influencing the size of the prokaryotic genome. In eukaryotic organisms, there is a paradox observed, namely that the number of genes that make up the genome does not correlate with genome size. In other words, the genome size is much larger than would be expected given the total number of protein coding genes.[11]
Genome size can increase by duplication, insertion, or polyploidization. Recombination can lead to both DNA loss or gain. Genomes can also shrink because of deletions. A famous example for such gene decay is the genome of Mycobacterium leprae, the causative agent of leprosy. M. leprae has lost many once-functional genes over time due to the formation of pseudogenes.[12] This is evident in looking at its closest ancestor Mycobacterium tuberculosis.[13] M. leprae lives and replicates inside of a host and due to this arrangement it does not have a need for many of the genes it once carried which allowed it to live and prosper outside the host. Thus over time these genes have lost their function through mechanisms such as mutation causing them to become pseudogenes. It is beneficial to an organism to rid itself of non-essential genes because it makes replicating its DNA much faster and requires less energy.[14]
An example of increasing genome size over time is seen in filamentous plant pathogens. These plant pathogen genomes have been growing larger over the years due to repeat-driven expansion. The repeat-rich regions contain genes coding for host interaction proteins. With the addition of more and more repeats to these regions the plants increase the possibility of developing new virulence factors through mutation and other forms of genetic recombination. In this way it is beneficial for these plant pathogens to have larger genomes.[15]
Mechanisms
Gene duplication
Gene duplication is the process by which a region of DNA coding for a gene is duplicated. This can occur as the result of an error in recombination or through a retrotransposition event. Duplicate genes are often immune to the selective pressure under which genes normally exist. As a result, a large number of mutations may accumulate in the duplicate gene code. This may render the gene non-functional or in some cases confer some benefit to the organism.[16][17]
Whole genome duplication
Similar to gene duplication, whole genome duplication is the process by which an organism's entire genetic information is copied, once or multiple times which is known as polyploidy.[18] This may provide an evolutionary benefit to the organism by supplying it with multiple copies of a gene thus creating a greater possibility of functional and selectively favored genes. However, tests for enhanced rate and innovation in teleost fishes with duplicated genomes compared with their close relative holostean fishes (without duplicated genomes) found that there was little difference between them for the first 150 million years of their evolution.[19]
In 1997, Wolfe & Shields gave evidence for an ancient duplication of the Saccharomyces cerevisiae (Yeast) genome.[20] It was initially noted that this yeast genome contained many individual gene duplications. Wolfe & Shields hypothesized that this was actually the result of an entire genome duplication in the yeast's distant evolutionary history. They found 32 pairs of homologous chromosomal regions, accounting for over half of the yeast's genome. They also noted that although homologs were present, they were often located on different chromosomes. Based on these observations, they determined that Saccharomyces cerevisiae underwent a whole genome duplication soon after its evolutionary split from Kluyveromyces, a genus of ascomycetous yeasts. Over time, many of the duplicate genes were deleted and rendered non-functional. A number of chromosomal rearrangements broke the original duplicate chromosomes into the current manifestation of homologous chromosomal regions. This idea was further solidified in looking at the genome of yeast's close relative Ashbya gossypii.[21] Whole genome duplication is common in fungi as well as plant species. An example of extreme genome duplication is represented by the Common Cordgrass (Spartina anglica) which is a dodecaploid, meaning that it contains 12 sets of chromosomes,[22] in stark contrast to the human diploid structure in which each individual has only two sets of 23 chromosomes.
Transposable elements
Transposable elements are regions of DNA that can be inserted into the genetic code through one of two mechanisms. These mechanisms work similarly to "cut-and-paste" and "copy-and-paste" functionalities in word processing programs. The "cut-and-paste" mechanism works by excising DNA from one place in the genome and inserting itself into another location in the code. The "copy-and-paste" mechanism works by making a genetic copy or copies of a specific region of DNA and inserting these copies elsewhere in the code.[23][24] The most common transposable element in the human genome is the Alu sequence, which is present in the genome over one million times.[25]
Mutation
Spontaneous mutations often occur which can cause various changes in the genome. Mutations can either change the identity of one or more nucleotides, or result in the addition or deletion of one or more nucleotide bases. Such changes can lead to a frameshift mutation, causing the entire code to be read in a different order from the original, often resulting in a protein becoming non-functional.[26] A mutation in a promoter region, enhancer region or transcription factor binding region can also result in either a loss of function, or an up or downregulation in the transcription of the gene targeted by these regulatory elements. Mutations are constantly occurring in an organism's genome and can cause either a negative effect, positive effect or neutral effect (no effect at all).[27][28]
Pseudogenes
Often a result of spontaneous mutation, pseudogenes are dysfunctional genes derived from previously functional gene relatives. There are many mechanisms by which a functional gene can become a pseudogene including the deletion or insertion of one or multiple nucleotides. This can result in a shift of reading frame, causing the gene to no longer code for the expected protein, introduce a premature stop codon or a mutation in the promoter region.[29] Often cited examples of pseudogenes within the human genome include the once functional olfactory gene families. Over time, many olfactory genes in the human genome became pseudogenes and were no longer able to produce functional proteins, explaining the poor sense of smell humans possess in comparison to their mammalian relatives.[30][31]
Exon shuffling
Exon shuffling is a mechanism by which new genes are created. This can occur when two or more exons from different genes are combined together or when exons are duplicated. Exon shuffling results in new genes by altering the current intron-exon structure. This can occur by any of the following processes: transposon mediated shuffling, sexual recombination or non-homologous recombination (also called illegitimate recombination). Exon shuffling may introduce new genes into the genome that can be either selected against and deleted or selectively favored and conserved.[32][33][34]
Genome reduction and gene loss
Many species exhibit genome reduction when subsets of their genes are not needed anymore. This typically happens when organisms adapt to a parasitic life style, e.g. when their nutrients are supplied by a host. As a consequence, they lose the genes needed to produce these nutrients. In many cases, there are both free living and parasitic species that can be compared and their lost genes identified. Good examples are the genomes of Mycobacterium tuberculosis and Mycobacterium leprae, the latter of which has a dramatically reduced genome.
Another beautiful example are endosymbiont species. For instance, Polynucleobacter necessarius was first described as a cytoplasmic endosymbiont of the ciliate Euplotes aediculatus. The latter species dies soon after being cured of the endosymbiont. In the few cases in which P. necessarius is not present, a different and rarer bacterium apparently supplies the same function. No attempt to grow symbiotic P. necessarius outside their hosts has yet been successful, strongly suggesting that the relationship is obligate for both partners. Yet, closely related free-living relatives of P. necessarius have been identified. The endosymbionts have a significantly reduced genome when compared to their free-living relatives (1.56 Mbp vs. 2.16 Mbp).[35]
Speciation

A major question of evolutionary biology is how genomes change to create new species. Speciation requires changes in behavior, morphology, physiology, or metabolism (or combinations thereof). The evolution of genomes during speciation has been studied only very recently with the availability of next-generation sequencing technologies. For instance, cichlid fish in African lakes differ both morphologically and in their behavior. The genomes of 5 species have revealed that both the sequences but also the expression pattern of many genes has quickly changed over a relatively short period of time (100,000 to several million years). Notably, 20% of duplicate gene pairs have gained a completely new tissue-specific expression pattern, indicating that these genes also obtained new functions. Given that gene expression is driven by short regulatory sequences, this demonstrates that relatively few mutations are required to drive speciation. The cichlid genomes also showed increased evolutionary rates in microRNAs which are involved in gene expression.[36][37]
Gene expression
Mutations can lead to changed gene function or, probably more often, to changed gene expression patterns. In fact, a study on 12 animal species provided strong evidence that tissue-specific gene expression was largely conserved between orthologs in different species. However, paralogs within the same species often have a different expression pattern. That is, after duplication of genes they often change their expression pattern, for instance by getting expressed in another tissue and thereby adopting new roles.[38]
Composition of nucleotides (GC content)
The genetic code is made up of sequences of four nucleotide bases: Adenine, Guanine, Cytosine and Thymine, commonly referred to as A, G, C, and T. The GC-content is the percentage of G & C bases within a genome. GC-content varies greatly between different organisms.[39] Gene coding regions have been shown to have a higher GC-content and the longer the gene is, the greater the percentage of G and C bases that are present. A higher GC-content confers a benefit because a Guanine-Cytosine bond is made up of three hydrogen bonds while an Adenine-Thymine bond is made up of only two. Thus the three hydrogen bonds give greater stability to the DNA strand. So, it is not surprising that important genes often have a higher GC-content than other parts of an organism's genome.[40] For this reason, many species living at very high temperatures such as the ecosystems surrounding hydrothermal vents, have a very high GC-content. High GC-content is also seen in regulatory sequences such as promoters which signal the start of a gene. Many promoters contain CpG islands, areas of the genome where a cytosine nucleotide occurs next to a guanine nucleotide at a greater proportion. It has also been shown that a broad distribution of GC-content between species within a genus shows a more ancient ancestry. Since the species have had more time to evolve, their GC-content has diverged further apart.
Evolving translation of genetic code
Amino acids are made up of three base long codons and both Glycine and Alanine are characterized by codons with Guanine-Cytosine bonds at the first two codon base positions. This GC bond gives more stability to the DNA structure. It has been hypothesized that as the first organisms evolved in a high-heat and pressure environment they needed the stability of these GC bonds in their genetic code.[41]
De novo origin of genes
Novel genes can arise from non-coding DNA. For instance, Levine and colleagues reported the origin of five new genes in the D. melanogaster genome from noncoding DNA.[42][43] Subsequently, de novo origin of genes has been also shown in other organisms such as yeast,[44] rice[45] and humans.[46] For instance, Wu et al. (2011) reported 60 putative de novo human-specific genes all of which are short consisting of a single exon (except one).[47] The probability of variations at given loci is likely to increase genesis of new genes. However, most mutations in general are deleterious to the cell, especially for genomes with high gene density, which are eventually lost in the purifying selection. However, non-coding regions such as the 'grounded' prophages are buffer zones which would tolerate variations thereby increasing the probability of de novo gene formation.[48] These grounded prophages and other such genetic elements are sites where genes could be acquired through horizontal gene transfer (HGT).
References
- Fiers W, Contreras R, Duerinck F, Haegeman G, Iserentant D, Merregaert J, et al. (April 1976). "Complete nucleotide sequence of bacteriophage MS2 RNA: primary and secondary structure of the replicase gene". Nature. 260 (5551): 500–7. Bibcode:1976Natur.260..500F. doi:10.1038/260500a0. PMID 1264203.
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. (February 2001). "The sequence of the human genome". Science. 291 (5507): 1304–51. Bibcode:2001Sci...291.1304V. doi:10.1126/science.1058040. PMID 11181995.
- Toussaint A, Chandler M (2012). "Prokaryote genome fluidity: toward a system approach of the mobilome". Bacterial Molecular Networks. Methods in Molecular Biology. 804. pp. 57–80. doi:10.1007/978-1-61779-361-5_4. ISBN 978-1-61779-360-8. PMID 22144148.
- Ruiz J, Pons MJ, Gomes C (September 2012). "Transferable mechanisms of quinolone resistance". International Journal of Antimicrobial Agents. 40 (3): 196–203. doi:10.1016/j.ijantimicag.2012.02.011. PMID 22831841.
- Koonin EV, Wolf YI (December 2008). "Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world". Nucleic Acids Research. 36 (21): 6688–719. doi:10.1093/nar/gkn668. PMC 2588523. PMID 18948295.
- Tortora, Gerard J. (2015). Microbiology: An Introduction. ISBN 978-0321929150.
- Dai L, Zimmerly S (October 2002). "The dispersal of five group II introns among natural populations of Escherichia coli". RNA. 8 (10): 1294–307. doi:10.1017/S1355838202023014. PMC 1370338. PMID 12403467.
- Blattner FR, Plunkett G, Bloch CA, Perna NT, Burland V, Riley M, et al. (September 1997). "The complete genome sequence of Escherichia coli K-12". Science. 277 (5331): 1453–62. doi:10.1126/science.277.5331.1453. PMID 9278503.
- International Human Genome Sequencing Consortium (October 2004). "Finishing the euchromatic sequence of the human genome". Nature. 431 (7011): 931–45. Bibcode:2004Natur.431..931H. doi:10.1038/nature03001. PMID 15496913.
- Gregory TR (February 2001). "Coincidence, coevolution, or causation? DNA content, cell size, and the C-value enigma". Biological Reviews of the Cambridge Philosophical Society. 76 (1): 65–101. doi:10.1017/S1464793100005595. PMID 11325054.
- Gregory TR (January 2002). "A bird's-eye view of the C-value enigma: genome size, cell size, and metabolic rate in the class aves". Evolution; International Journal of Organic Evolution. 56 (1): 121–30. doi:10.1111/j.0014-3820.2002.tb00854.x. PMID 11913657.
- Singh P, Cole ST (January 2011). "Mycobacterium leprae: genes, pseudogenes and genetic diversity". Future Microbiology. 6 (1): 57–71. doi:10.2217/fmb.10.153. PMC 3076554. PMID 21162636.
- Eiglmeier K, Parkhill J, Honoré N, Garnier T, Tekaia F, Telenti A, et al. (December 2001). "The decaying genome of Mycobacterium leprae". Leprosy Review. 72 (4): 387–98. doi:10.5935/0305-7518.20010054. PMID 11826475.
- Rosengarten R, Citti C, Glew M, Lischewski A, Droesse M, Much P, et al. (March 2000). "Host-pathogen interactions in mycoplasma pathogenesis: virulence and survival strategies of minimalist prokaryotes". International Journal of Medical Microbiology. 290 (1): 15–25. doi:10.1016/S1438-4221(00)80099-5. PMID 11043978.
- Raffaele S, Kamoun S (May 2012). "Genome evolution in filamentous plant pathogens: why bigger can be better". Nature Reviews. Microbiology. 10 (6): 417–30. doi:10.1038/nrmicro2790. PMID 22565130.
- Zhang, Jianzhi (2003). "Evolution by gene duplication: an update". Trends in Ecology & Evolution. 18 (6): 292–298. doi:10.1016/S0169-5347(03)00033-8.
- Taylor JS, Raes J (2004). "Duplication and divergence: the evolution of new genes and old ideas". Annual Review of Genetics. 38: 615–43. doi:10.1146/annurev.genet.38.072902.092831. PMID 15568988.
- Song C, Liu S, Xiao J, He W, Zhou Y, Qin Q, Zhang C, Liu Y (April 2012). "Polyploid organisms". Science China Life Sciences. 55 (4): 301–11. doi:10.1007/s11427-012-4310-2. PMID 22566086.
- Clarke JT, Lloyd GT, Friedman M (October 2016). "Little evidence for enhanced phenotypic evolution in early teleosts relative to their living fossil sister group". Proceedings of the National Academy of Sciences of the United States of America. 113 (41): 11531–11536. doi:10.1073/pnas.1607237113. PMC 5068283. PMID 27671652.
- Wolfe KH, Shields DC (June 1997). "Molecular evidence for an ancient duplication of the entire yeast genome". Nature. 387 (6634): 708–13. Bibcode:1997Natur.387..708W. doi:10.1038/42711. PMID 9192896.
- Dietrich FS, Voegeli S, Brachat S, Lerch A, Gates K, Steiner S, Mohr C, Pöhlmann R, Luedi P, Choi S, Wing RA, Flavier A, Gaffney TD, Philippsen P (April 2004). "The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome". Science. 304 (5668): 304–7. Bibcode:2004Sci...304..304D. doi:10.1126/science.1095781. PMID 15001715.
- Buggs RJ (November 2012). "Monkeying around with ploidy". Molecular Ecology. 21 (21): 5159–61. doi:10.1111/mec.12005. PMID 23075066.
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, SanMiguel P, Schulman AH (December 2007). "A unified classification system for eukaryotic transposable elements". Nature Reviews Genetics. 8 (12): 973–82. doi:10.1038/nrg2165. PMID 17984973.
- Ivics Z, Izsvák Z (January 2005). "A whole lotta jumpin' goin' on: new transposon tools for vertebrate functional genomics". Trends in Genetics. 21 (1): 8–11. doi:10.1016/j.tig.2004.11.008. PMID 15680506.
- Oler AJ, Traina-Dorge S, Derbes RS, Canella D, Cairns BR, Roy-Engel AM (June 2012). "Alu expression in human cell lines and their retrotranspositional potential". Mobile DNA. 3 (1): 11. doi:10.1186/1759-8753-3-11. PMC 3412727. PMID 22716230.
- Griffiths A (December 2011). "Slipping and sliding: frameshift mutations in herpes simplex virus thymidine kinase and drug-resistance". Drug Resistance Updates. 14 (6): 251–9. doi:10.1016/j.drup.2011.08.003. PMC 3195865. PMID 21940196.
- Eyre-Walker A, Keightley PD (August 2007). "The distribution of fitness effects of new mutations". Nature Reviews Genetics. 8 (8): 610–8. doi:10.1038/nrg2146. PMID 17637733.
- Gillespie JH (September 1984). "Molecular Evolution Over the Mutational Landscape". Evolution; International Journal of Organic Evolution. 38 (5): 1116–1129. doi:10.2307/2408444. JSTOR 2408444. PMID 28555784.
- Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR (May 2011). "Pseudogenes: pseudo-functional or key regulators in health and disease?". RNA. 17 (5): 792–8. doi:10.1261/rna.2658311. PMC 3078729. PMID 21398401.
- Sharon D, Glusman G, Pilpel Y, Horn-Saban S, Lancet D (November 1998). "Genome dynamics, evolution, and protein modeling in the olfactory receptor gene superfamily". Annals of the New York Academy of Sciences. 855 (1): 182–93. Bibcode:1998NYASA.855..182S. doi:10.1111/j.1749-6632.1998.tb10564.x. PMID 9929603.
- Mombaerts P (2001). "The human repertoire of odorant receptor genes and pseudogenes". Annual Review of Genomics and Human Genetics. 2: 493–510. doi:10.1146/annurev.genom.2.1.493. PMID 11701659.
- Liu M, Grigoriev A (September 2004). "Protein domains correlate strongly with exons in multiple eukaryotic genomes--evidence of exon shuffling?". Trends in Genetics. 20 (9): 399–403. doi:10.1016/j.tig.2004.06.013. PMID 15313546.
- Froy O, Gurevitz M (December 2003). "Arthropod and mollusk defensins--evolution by exon-shuffling". Trends in Genetics. 19 (12): 684–7. doi:10.1016/j.tig.2003.10.010. PMID 14642747.
- Roy SW (July 2003). "Recent evidence for the exon theory of genes". Genetica. 118 (2–3): 251–66. doi:10.1023/A:1024190617462. PMID 12868614.
- Boscaro V, Felletti M, Vannini C, Ackerman MS, Chain PS, Malfatti S, Vergez LM, Shin M, Doak TG, Lynch M, Petroni G (November 2013). "Polynucleobacter necessarius, a model for genome reduction in both free-living and symbiotic bacteria". Proceedings of the National Academy of Sciences of the United States of America. 110 (46): 18590–5. Bibcode:2013PNAS..11018590B. doi:10.1073/pnas.1316687110. PMC 3831957. PMID 24167248.
- Brawand D, Wagner CE, Li YI, Malinsky M, Keller I, Fan S, et al. (September 2014). "The genomic substrate for adaptive radiation in African cichlid fish". Nature. 513 (7518): 375–381. Bibcode:2014Natur.513..375B. doi:10.1038/nature13726. PMC 4353498. PMID 25186727.
- Jiggins CD (September 2014). "Evolutionary biology: Radiating genomes". Nature. 513 (7518): 318–9. Bibcode:2014Natur.513..318J. doi:10.1038/nature13742. PMID 25186726.
- Kryuchkova-Mostacci N, Robinson-Rechavi M (December 2016). "Tissue-Specificity of Gene Expression Diverges Slowly between Orthologs, and Rapidly between Paralogs". PLoS Computational Biology. 12 (12): e1005274. doi:10.1371/journal.pcbi.1005274. PMC 5193323. PMID 28030541.
- Li W (November 2011). "On parameters of the human genome". Journal of Theoretical Biology. 288: 92–104. doi:10.1016/j.jtbi.2011.07.021. PMID 21821053.
- Galtier N (February 2003). "Gene conversion drives GC content evolution in mammalian histones". Trends in Genetics. 19 (2): 65–8. doi:10.1016/S0168-9525(02)00002-1. PMID 12547511.
- Šmarda P, Bureš P, Horová L, Leitch IJ, Mucina L, Pacini E, et al. (September 2014). "Ecological and evolutionary significance of genomic GC content diversity in monocots". Proceedings of the National Academy of Sciences of the United States of America. 111 (39): E4096-102. doi:10.1073/pnas.1321152111. PMC 4191780. PMID 25225383.
- Levine MT, Jones CD, Kern AD, Lindfors HA, Begun DJ (June 2006). "Novel genes derived from noncoding DNA in Drosophila melanogaster are frequently X-linked and exhibit testis-biased expression". Proceedings of the National Academy of Sciences of the United States of America. 103 (26): 9935–9. Bibcode:2006PNAS..103.9935L. doi:10.1073/pnas.0509809103. PMC 1502557. PMID 16777968.
- Zhou Q, Zhang G, Zhang Y, Xu S, Zhao R, Zhan Z, Li X, Ding Y, Yang S, Wang W (September 2008). "On the origin of new genes in Drosophila". Genome Research. 18 (9): 1446–55. doi:10.1101/gr.076588.108. PMC 2527705. PMID 18550802.
- Cai J, Zhao R, Jiang H, Wang W (May 2008). "De novo origination of a new protein-coding gene in Saccharomyces cerevisiae". Genetics. 179 (1): 487–96. doi:10.1534/genetics.107.084491. PMC 2390625. PMID 18493065.
- Xiao W, Liu H, Li Y, Li X, Xu C, Long M, Wang S (2009). El-Shemy HA (ed.). "A rice gene of de novo origin negatively regulates pathogen-induced defense response". PLOS ONE. 4 (2): e4603. Bibcode:2009PLoSO...4.4603X. doi:10.1371/journal.pone.0004603. PMC 2643483. PMID 19240804.
- Knowles DG, McLysaght A (October 2009). "Recent de novo origin of human protein-coding genes". Genome Research. 19 (10): 1752–9. doi:10.1101/gr.095026.109. PMC 2765279. PMID 19726446.
- Wu DD, Irwin DM, Zhang YP (November 2011). "De novo origin of human protein-coding genes". PLoS Genetics. 7 (11): e1002379. doi:10.1371/journal.pgen.1002379. PMC 3213175. PMID 22102831.
- Ramisetty BC, Sudhakari PA (2019). "Bacterial 'Grounded' Prophages: Hotspots for Genetic Renovation and Innovation". Frontiers in Genetics. 10: 65. doi:10.3389/fgene.2019.00065. PMC 6379469. PMID 30809245.